以前我一直希望人类基因组的序列在完全解开后,会有72415个基因。我之所以特别看重这个数目,跟人类基因组计划第一个惊人的发现有关。1999年12月,第一个完成定序的染色体是位于两大定序里程碑(10亿和20亿个碱基序列)之间的第22号染色体。尽管它很小,在整个基因组中只占1.1%,但仍长达3340万个碱基对。由小窥大,这是我们首次对基因组的全貌有概括的了解;如同《自然》杂志的评论家所言,这就像“第一次看到新星球的表面或地景”。最有趣的是染色体上基因的密度。以第22号染色体作为整个基因组的缩影,应该是很合理的,所以照理说我们会在它的序列里发现1.1%的人类基因、换句话说,教科书上估计人类基因的总数约为10万个,照这样推算,第22号染色体应该大约有1100个基因。但是我们却只在它上面发现545个基因。这似乎强烈暗示,人类基因组不像我们原先所以为的有那么多基因。
人类的基因数目突然成为热门话题。2000年5月,在冷泉港实验室一场有关基因组的会议上,来自桑格中心,负责以计算机分析基因组序列的伯尼(Ewan Birney),设下他称之为Genesweep的赌局。这场赌局赌的是人类的基因总数,等2003年完成定序时就可以知道大概的正确数字,届时谁猜的数目最接近,谁就是优胜者。伯尼会成为人类基因组计划的地下赌注经纪人一点也不奇怪,因为数字正是他的专长。从伊顿公学毕业后,他借住我位于长岛的房子,花了一年时间解决生物学上的定量问题。英国年轻人在高中毕业后上大学前的那一年,多半会选择到喜马拉雅山旅行或是到里约热内卢的酒吧打工,但他的选择大不相同。伯尼在冷泉港实验室工作时就写出两篇重要的研究论文,那时他甚至还没进牛津呢。

后基因组时代:基因启闭模式的微阵列(microarray)分析。在这个图例中,每个光点对应疟原虫6000个不同基因之一,疟原虫会引起最严重的一种疟疾。我们在搜寻疫苗或解药时,必须知道在生命周期的不同阶段,哪些基因是活跃的。红点显示这个基因在一个阶段是活跃的,但在另一个阶段则不活跃,绿点则与之相反。在两个阶段都活跃的基因一般会以黄点呈现。
伯尼最初要求的赌资是1美元,但是随着让我们更接近最终数字的预测数目一发表,赌资也跟着提高。我从一开始就加入,所以只花了1美元押在72415上。这个数目可不是乱选的,我以第22号染色体的结果为本,考虑教科书10万个的说法和当时的预测值5万个后,才折中地选了它。
或许惟一会跟基因数一样引起这么多无聊臆测的问题,是我们所定序的究竟是谁的基因。原则上,这是机密数据,所以拿这个来打赌的话,大概不会有输赢。就公共人类基因组计划来说,我们定序的DNA样本来自纽约州布法罗附近一些随机选出的人,样本的处理也是在相同的地区进行。所谓处理是先分离出DNA,再殖入人造细菌染色体,以便建立图谱与定序。起初赛雷拉基因公司宣称,他们的材料也是取自六个匿名捐赠、来自多种文化的人,但是在2002年,温特忍不住把谜底公诸于世,宣称他们定序的主要基因组其实是他的。如今,那个序列成了温特跟赛雷拉之间仅余的联结。虽然人类基因组定序很吸引人,又有新闻价值,但是看来并没有什么商业效益,因此赛雷拉后来转型为制药公司,并在2002年和它的创办人分道扬镳。温特又成立了两家新公司,一家研究现代遗传学引起的道德议题,另一家利用细菌基因组来寻找新的再生能源。
在人类基因组图谱出炉后,已证实第22号染色体的基因密度并没什么出奇之处。其实,以它的大小小来说,拥有545个基因还算蛮多的。大小跟它差不多的第21号染色体,只找出236个基因。根据现在的估计,在人类的全套24条染色体(22条+X+Y)上,基因总数也只不过2万多个。当然,寻找基因的工作还没结束,我们还会发现更多基因,但基因总数绝对远在3万以下,距离从前教科书说的10万就更遥远了。
基因总数究竟会是多少?我们只能等着瞧。事实上,寻找基因不是这么简单:蛋白质编码区全是由A,T,G,C构成的链,而且这些链深埋在基因组其他的A,T,G,C之间,一点也不明显。我们也必须记得,人类基因组大约只有2%为蛋白质编码,至于那些被称为“垃圾”的部分,则是由显然不具功能、长短不一的片段所构成,其中还有许多会重复出现。就连基因本身也含有垃圾片段;在有许多非编码片段(即插入序列)的情况下,基因有时占有一长段绵延的DNA,而编码片段就像是分子高速公路上一段段荒凉道路之间零星、孤立的城镇。目前已知最长的人类基因是肌萎缩蛋白(dystrophin,它发生突变时会引起肌肉萎缩症),长达240万个碱基对,其中只有11055个(仅占基因的0.5%)为蛋白质编码,其余部分构成这个基因的79个插入序列(典型的人类基因有8个插入序列)。基因辨识工作之所以困难,原因就在于基因组这种庞杂的结构。
在我们对老鼠基因比较了解后,要寻找人类基因已没有那么棘手,这都要归功于进化:如同所有哺乳动物的基因组,人类和老鼠的基因组中具有功能的部分相当类似,从人与鼠在远古的共同祖先一直到现在,这些部分并未产生太大的歧异。相对之下,那些由垃圾DNA组成的部分向来走在进化的最前端;由于不像编码片段有自然选择的监督,因此垃圾区累积了大量突变,人与鼠在遗传上的歧异程度也以这些区段为最高。因此寻找人与鼠在基因组序列中的相似部分,成了辨识基因具有功能的区段的有效方法。
完成河豚基因组的草图也有助于辨识人类基因。这种日本老饕最爱的鱼含有强烈的神经毒素;老练的厨师会先把含有毒素的器官移除,所以食客只会觉得嘴里有点麻而已。但是每年大约有80人因河豚的制作过程不周而死亡,因此日本法律明文禁止皇室享受这道美食。10多年前,布雷纳开始“爱上”河豚,至少是把它们当做研究基因的对象。河豚基因组的规模只有人类的1/9,但所含的垃圾区段少得多:它的基因组中约1/3负责为蛋白质编码。在布雷纳的领导下,河豚基因组的草图只花了1200万美元左右的经费就完成了,以基因组定序的标准来说,算是很便宜。目前看来,河豚基因数大约在3.2万到4万之间,和人类差不多。有趣的是,河豚基因的插入序列数目虽然跟人类及老鼠基因的差不多,但长度通常短得多。

人类第2号色体染色体上的基因:2.55亿个碱基对。
根据目前的估计,人类基因数大约是在3.5万(注:原书如此,现在一般认为是在20000-25000之间)左右,即使基因数已大幅向下修正,但对一般人来说,光看这个数字可能会给他们一种错觉,有点夸大了我们基因的复杂程度。在进化过程中,某些基因会衍生出一组相关的基因,形成一群功能类似、但有细微差异的基因。这些所谓的“基因家族”(gene family)完全是意外的产物:在制造卵细胞或精细胞的过程中,某染色体的一个区段无意间遭到复制,使得这个染色体上的某个特定基因多了一个复本。只要其中一个基因能发挥功能,自然选择就不会去检查另一个,而随着突变不断累积,这个额外的基因可能走上歧异的进化道路。偶尔这些突变会造成基因获得新的功能,通常是与原基因密切相关的功能。人类基因所负责的“主题”,种类并不太多;事实上,我们许多基因的主题都一样,只是略有变化而已。例如我们有575个基因负责编码不同形式的蛋白质激酶(protein kinase enzyme),亦即在细胞周围传递讯号的化学使者。大约有900个人类基因让我们有嗅闻的能力:它们编码的蛋白质是气味受体,每种受体辨识一种不同气味的分子或一整类分子。这900个侦察气味的基因大体上也存在于老鼠体内,但其中有个差异:老鼠已经适应以夜行为主的生活,对嗅觉的需要程度较大——自然选择筛选出嗅觉比较好的老鼠,并让这900个气味基因中的大多数持续运作。然而,人类的这些基因大约有60%已经在进化过程中退化。可能的原因是:当我们对视觉的依赖增加时,我们便不再需要那么多嗅觉受体,所以当突变造成许多嗅觉基因无法制造正常的蛋白质,使我们的嗅觉变得比其他恒温动物差时,自然选择并没有插手。
那么,我们的基因数跟其他生物的基因数相比如何?
就基因的总量而言,我们也就比杂草类的植物多一点点而已!跟线虫比,就更令人吃惊。线虫只有959个细胞(人类估计约有100兆个细胞),其中302个是神经细胞,构成线虫极度简单的脑(我们的脑有1000亿个神经细胞),我们和线虫在结构的复杂度上有天壤之别,但我们的基因总数还不到线虫的两倍。我们要怎么解释这令人困窘的数字?其实我们一点也不必不好意思:看来我们人类就是能用这么一套遗传硬件来做更多的事。
事实上,我认为智力与低基因数之间有一种相互关系。我的看法是:所谓聪明,就是像我们或果蝇一样,拥有一个相当不错的神经中心,让我们能以相对很少的基因,执行复杂的机能。(把几万个称为“很少”,好像有点怪,不过这是相较而言。)脑赋予我们的感觉能力与神经操控肌肉运动的能力,远超过没有眼睛、移动缓慢的线虫,因此我们可以选择的行为反应,范围大得多。至于不能移动的植物,选择更少,它们得用上所有的遗传资源才能应付环境中的偶发事件。相对地,聪明的物种在遇到天气骤然变冷等偶发事件时,可以运用神经细胞去寻找更适合的环境(例如温暖的洞穴)。

老鼠与人类同一个基因的DNA比较图,包括一个插入序列(基因非编码的区域,如黑框所示)和两个表现序列的一部分(这些区域为该基因制造的蛋白质编码)。黄色碱基是人和鼠的序列历经进化而一直没有改变的部分,短橫线代表该物种在那个位置失去一个碱基,老鼠和人类序列的相似性显示,自然选择在消除突变上非常有效,在插入序列中,突变通常无所谓,而在表规序列中的突变则有可能会破坏蛋白质的功能。我们可以看到人类和老鼠在插入序列上的差异比表现序列多得多。
脊椎动物也可能因为基因开关系统而变得更加复杂。基因开关一般位于基因附近,在基因组定序的工作完成后,我们现在可以仔细分析这些基因旁侧的区域了。基因调节作用就是在这些区域发生的,由接合至DNA的调节蛋白质(reguatory protein)来启动或关闭邻近的基因。相较于比较简单的生物,控制脊椎动物基因的启闭机制似乎要复杂得多。脊推动物之所以这么复杂,正是因为基因之间复杂精巧的协调作用。此外,一种特定基因有可能额外产生许多不同的蛋白质,这或者是因为不同的表现序列耦合在一起,从而创造出稍有不同的蛋白质(此过程称为替代性剪接[clternative splicing]),不然就是因为蛋白质在制造后发生了生化上的变化。
人类基因数出乎意料地少,引起一些人在报刊的言论版撰文讨论其意义,它们的主旨大多相同。进化生物学家古尔德(Stephen Jay Gould,他不幸于2002年病逝,激昂言论从此绝响)在《纽约时报》发表文章,认定低基因数为还原论(reductionism)敲响了丧钟。还原论几乎是所有的生物学研究遵奉的教条,这个理论认为一个复杂的系统是由下往上建立的,换句话说,要深入了解一个组织的复杂层次,就必须先了解这个组织比较简单的层次,再把这些比较简单的动态情况组合起来。根据这个看法,只要了解基因组的运作,我们最终必然可以了解生物体是如何组成的。古尔德等人以基因数少得惊人这一点作为证据,证明这种由下而上的方法不仅难以实行,也根本是无效的。有鉴于人类基因组比预想中简单,反还原论者认为人类这个生物体就是活生生的证据,证明要了解我们自身,不是光靠把所有较小的过程相加就可以。对他们而言,人类的基因数少,正暗示着我们每一个人的主要决定因子是教养(后天因素),而不是天性(先天因素)。简言之,这等于一篇独立宣言,宣示我们人类不是像原先所想的,完全受基因支配。
我跟古尔德一样,也了解教养在我们每一个人的塑造过程中很重要,但他在评估天性的角色上却完全错误:我们的基因数低,并不能证明对生物系统采取还原论的方法是无效的,也不能在逻辑推论上证明我们不是由基因所决定的。包含黑猩猩基因组的受精卵必定会孕育出黑猩猩,而含有人类基因组的受精卵只会孕育出人类。无论接触多少古典音乐或暴力的电视画面,都不会改变这个事实。没错,我们还要许久的时间,才能充分了解这两个极为类似的基因组中所含的资料是怎么被运用的,为何能制造出两种显然极为不同的生物;但是,最后会形成哪种生物,其绝大部分的指令早就写在每个细胞里,亦即基因组中,这是不变的事实。事实上,我认为在以还原论的方法来研究生物学上,人类基因数少,反而是好消息:要理清3.5万个基因的影响,要比理清10万个简单得多。
我们人类的基因数或许不是很多,但是就如同芜蔓的肌萎缩蛋白基因所显示的,我们拥有一个庞大杂乱的基因组。我们可以再度拿线虫作比较:我们的基因数还不到线虫的两倍,但基因组的体积却大了33倍。为何会有这样的差异?建立基因图谱的专家把人类的基因组描述为一个偶尔有绿洲(即基因)的沙漠。在基因组中,有50%是不具明显功能、垃圾般的重复序列;我们的DNA足足有10%是由分散在各处的100万个单一序列复本所构成,这个序列称为Alu(节杆菌Arthrobacter luteus的缩写,因为这个序列最初是在这种细菌中被分离出来):
GGCCGGGCGCGGTGGCTCACGCCTGTAATCCCAGCACTTTGG
GAGGCCGAGGCGGGCGGATCACCTGAGGTCAGGAGTTCGAGA
CCAGCCTGGCCAACATGGTGAAACCCCGTCTCTACTAAAAATA
CAAAAATTAGCCGGGCGTGGTGGCGCGCGCCTGTAATCCCAG
CTACTCGGGAGGCTGAGGCAGGAGAATCGCTTGAACCCGGGA
GGCGGAGGTTGCAGTGAGCCGAGATCGCGCCACTGCACTCCA
GCCTGGGCGACAGAGCGAGACTCCGTCTCAAAAAA
把这个序列写100万次,就可以了解Alu序列在我们的DNA中占了多大的规模。事实上,重复序列的数目甚至比表面上看来还多:一度可以立即辨识的重复序列,在经过许多世代的突变后,产生的歧异已经大到让人看不出它们原本是某类重复DNA的成员了。假设现在有三个短的重复序列:ATTGATTGATTG。一段时间后,突变会改变它们,如果这段期间很短,我们仍可看出它们的来源:ACTGATGGGTTG。但是,如果时间较长,它们的原始身份就会在混杂的突变中完全消失:ACCTCGGGGTCG。在许多其他物种中,重复DNA的比例低得多:芥菜种子的基因组只有11%是重复序列,线虫是7%,果蝇只有3%。我们人类的基因组体积大,主要就是因为包含的垃圾序列比其他许多物种多。
生物在垃圾DNA数量上的差异,解释了长期以来的一个进化之谜。一般的预期是:复杂生物的基因组会比简单生物的大,因为它们必须编码更多的信息。基因组的大小和生物个体的复杂程度,的确有相互关系,例如酵母菌的基因组比大肠杆菌的基因组大,但比我们人类的小。然而,这个相互关系相当薄弱。

头顶的洋葱:这些洋葱的基因组是洋葱小贩的基因组的六倍。
自然选择会尽量使基因组变小,这是合理的假设。毕竟,每次细胞分裂时,它都必须复制所有的DNA;必须复制的内容愈多,出错机会愈大,这个过程所需要的能量与时间也愈多。这对阿米巴原虫、蝾螈或肺鱼(lungfish)都是相当辛苦的工作。既然如此,这些物种的DNA数量为何会失控至此?就这些异常大的基因组而言,我们只能推论还有其他的选择力量介入,抵消了保持小基因组的向然选择推力。比方说,对有可能暴露在极端环境的生物来说,基因组大或许比较有利。肺鱼住在陆地与水的交界处,可以借由把自己埋在泥巴里来度过漫长的干旱期;它们需要的基因硬件有可能比适应单一环境的生物来得多。
有两个主要的进化机制可以解释这种DNA过多的观象:基因组加倍(genome doubling),以及一个基因组内特记序列的增殖(proliferation)。许多物种,尤其是植物,其实是两个先前存在的物种互相杂交的结果。这些新物种通常只是结合了两个亲代的全套DNA,从而创造出双倍基因组。或者也有可能因为某种遗传意外,使一个基因组在没有其他物种的输入之下倍增。以面包酵母为例,它大约有6000个基因,但进一步的检视显示,这些基因大部分是重复的,酵母的许多基因经常有两个不同的复本。在酵母进化史的某个早期阶段,它的基因组显然倍增了。起初这些基因复本必定是一模一样的,但是在长时间后,它们已经产生歧异。
另一个更丰富的多余DNA来源是:能自行复制并插入基因组中多个位置的基因序列发生增殖。现在已经发现各种各样“可移动遗传因子”(mobile element)。但是1950年麦克林托克(Barbara McClintock)首次发表“跳跃基因”(jumping gene)的概念时,习于孟德尔那套简单逻辑的科学家大多无法接受。麦克林托克是一位卓越的玉米遗传学家,但研究生涯相当坎坷。1941年,她未能拿到密苏里大学的永久教职后,来到冷泉港实验室,后来一直是这里的活跃成员,直到1992年以90岁高龄辞世为止。她曾经告诉一位同事说,“一定要相信自己眼中所见。”这就是她研究科学的方式:她认为一些遗传因子会在基因组中移动的革命性构想,完全得自可观察的事实。她研究决定玉米粒颜色的遗传机制,发现有时个别玉米粒发育到一半时,颜色会变换,一颗玉米粒可能会变成杂色,有预期中的黄色细胞,也有紫色细胞。要怎么解释这种突然的转换?麦克林托克推论,这是因为有可移动的遗传因子在色素基因里跳进跳出。

可移动遗传因子的发现者麦克林托克最初提出这个概念时备受奚落;直到30年后,才在1983年获得诺贝尔奖。
惟有在重组DNA技术问世后,我们才了解可移动因子有多常见;现在我们已经知道,它们就算不是大多数基因组,也是许多基因组的主要成分,包括人类基因组在内。这些可移动因子在相同基因组的不同位置一再出现,其中有些最常见的可移动因子所取的名字,反应出其四处移动的存在方式,例如有两个果蝇的可移动因子取名为“吉普赛”(gypsy)和“流浪工人”(hobo)。有人在研究团藻(volvox)这种简单植物时,发现其中一个可移动因子在基因组四周跳跃的能力特别强,因而称之为“(麦克)乔丹因子”([Michael]Jordan element)。
可移动因子包含为一些酶编码的DNA序列,这些酶有剪贴染色体DNA的能力,其功能就是要确保可移动因子的复本会插入染色体上的新位置。如果一个可移动因子跳跃至一个垃圾序列,生物体的功能不会受影响,惟一的结果是更多的垃圾DNA。但是,如果一个可移动因子跳至重要的基因,从而使这个基因丧失功能,则自然选择就会介入:这个生物体可能死亡或因为不能生育等因素,无法将这个含有跳跃因子的新基因传递下去。可移动因子的移动鲜少会创造新基因或造成对原生物体有利的改变。因此,经过长久的进化过程后,可移动因子的影响似乎主要在于产生“新奇性”,例如杂色的玉米粒。奇怪的是,在最近的人类史上,极少有积极跳跃的遗传因子之证据:我们的垃圾DNA似乎大多是在许久之前所产生的。相反地,老鼠基因组含有许多积极跳跃的可移动因子,但这似乎没有对老鼠造成太多困扰;老鼠的繁殖能力原本就高,这可能使他们比较能够容忍经常有因子跳至功能极重要的基因区所带来的遗传灾难。
我们曾借由大肠杆菌了解许多有关DNA如何发挥功能的基本信息,它作为一种模型生物的贡献真是无可比拟,难怪人类基因组计划把破解它的基因组列为优先要务之一。威斯康辛大学的布莱特纳(Fred Blattner)是最急于开始定序大肠杆菌的人,但他申请补助的提案没有下文,直到人类基因组计划获得经费后,才拨给他一笔可观的研究费。要不是他最初不愿釆取自动化定序法,他的实验室会是最早找出一种细菌基因组完整序列的。但是在1991年,他扩大研究规模的策略却是釆取了传统方法:雇用更多的大学生。另一位同样较晚利用自动化的人是吉尔伯特,我早在两年前就敦促他定序当时已知最小的细菌“支原体”(mycoplasma)的基因组,这种微小细菌以细胞为家。吉尔伯特想采取一种新的人工定序策略,遗憾的是,当这个策略无法成功时,支原体计划也跟着结束。然而,布莱特纳最后仍及时采取了自动化定序法,并在1997年发现大肠杆菌基因组包含大约4100个基因。
但是率先定序任一细菌之基因组的比赛,早在两年前就已经由基因组研究所(TIGR)拔得头筹,优胜者是由史密斯(Hamilton Smith)、温特和他太太佛瑞瑟(Claire Fraser)率领的庞大团队。他们定序的对象是流感嗜血杆菌。在更早的20年前,史密斯(他身高6.6英尺,原本主修数学,后来改念医学院)就已经从这种细菌分离出第一个有用的DNA限制酶,而这个功绩也让他赢得1978年的诺贝尔生理医学奖。他们这个团队分工是由史密斯制备流感嗜血杆菌的DNA,然后由温特和佛瑞瑟用全基因组随机定序法(WGS)去定序这种杆菌的180万个碱基对。光是记录这第一个“小”基因组,就足以让人知道那些等着被定序、体积更大的基因组有多可观:如果把流感嗜血杆菌基因组所有的A、T、G和C,都印在跟本书书页同样大小的纸张上,可能要4000页左右才印得完。它一共有1727个基因,每个基因平均要用掉两页。在这些基因当中,只有55%的功能可以辨识:例如制造能量至少涉及112个基因,而DNA的复制、修补与重组至少需要87个基因。我们可以从它们的序列中看出,剩余45%的基因也都有功能,只是在现阶段还无法确认这些功能是什么。
就细菌的标准而言,流感嗜血杆菌的基因组相当小。细菌基因组的大小与此特定菌种可能遭遇的环境多样性有关。在毫无变化的单一环境(例如另一种生物的肠道)过着单调的生活,这个物种只需要相对较小的基因组就过得去。然而,如果它想看看广大的世界,就比较容易遇到变化更多的情况,这时它必须具备足以应对环境的条件,而这种具有弹性的应对能力通常来自可供替换的好几组基因,每组都适合特定的情况,而且随时可以启动。
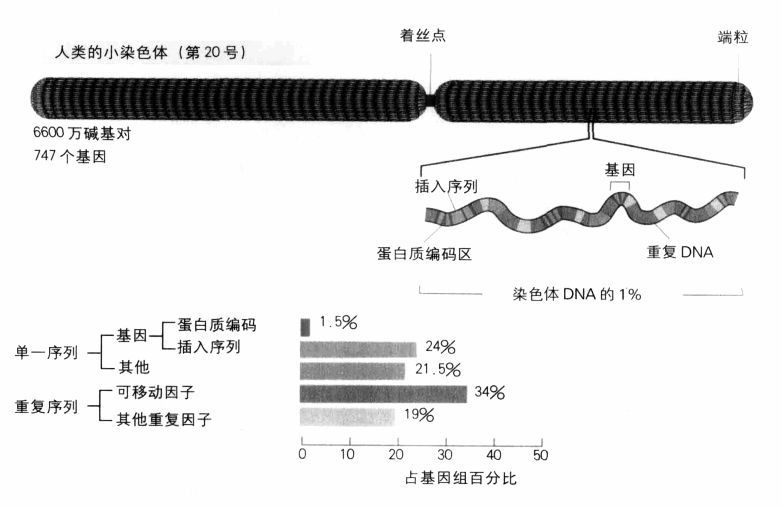
人类基因组的面貌:一个小染色体(第20号)的主要特征。
锈色假单孢菌(pseudomonas aeruginosa,亦称绿脓杆菌)是一种会在人体内引起感染的细菌(对纤维囊泡症病患者特别危险),它可以在许多不同的环境中存活。我们在第五章中看到,经过基因改造的一个相关菌种成为第一个被申请专利的活生物,在那个例子中,细菌适应了油膜里的生活,而油膜是跟人的肺脏相当不同的环境。锈色假单孢菌基因组包含640万个碱基对,及5570个基因。在这些基因中,大约7%编码转录因子,即可以开启和关闭基因的蛋白质;因此这种菌的所有基因中,有相当可观的比例是用于调节机制。在20世纪60年代初,莫诺和雅各布曾预测过大肠杆菌“抑制子”的存在(见第三章),这个抑制子正是这类转录因子。因此,我们获得一条经验法则:一个菌种可能遇到的各种环境愈多,它的基因组就愈大,而该基因组中用于基因启闭的比例也愈高。
基因组研究所并没有在做完流感嗜血杆菌后就歇手。1995年,在与北卡罗来纳大学的哈奇森(Clyde Hutchison)合作下,这家研究机构在当时戏称为“最小基因组计划”的研究中,定序了生殖道支原体(Mycoplasma genitalium)基因组。生殖道支原体(虽然名称有点不祥的味道,但它在人类排泄道中是无害的生物)拥有目前已知最小的非病毒基因组,约有58万碱基对。(病毒的基因组更小,但是它们可以利用宿主的各种基因,所以就算本身没有许多基本功能所需的基因资源,但仍能生存。)后来发现这个相对较短的支原体序列包含517个基因。大家自然会问一个问题:这是维持生命所需最少的基因数吗?后续的研究是破坏掉生殖道支原体的某些基因,看哪些是维持生命所必需的,哪些不是。目前看来,维持生命的最小基因组所包含的基因不超过350个,甚至可能少至260个。无可否认地,这个“最小”多少是人为定义的,因为,即使破坏细菌的一些基因,实验所用的生长培养基仍能提供这些虚弱的细菌生存所需要的一切物质。这有点像宣称肾脏不是生命所需,因为病人可以靠透析仪生存。
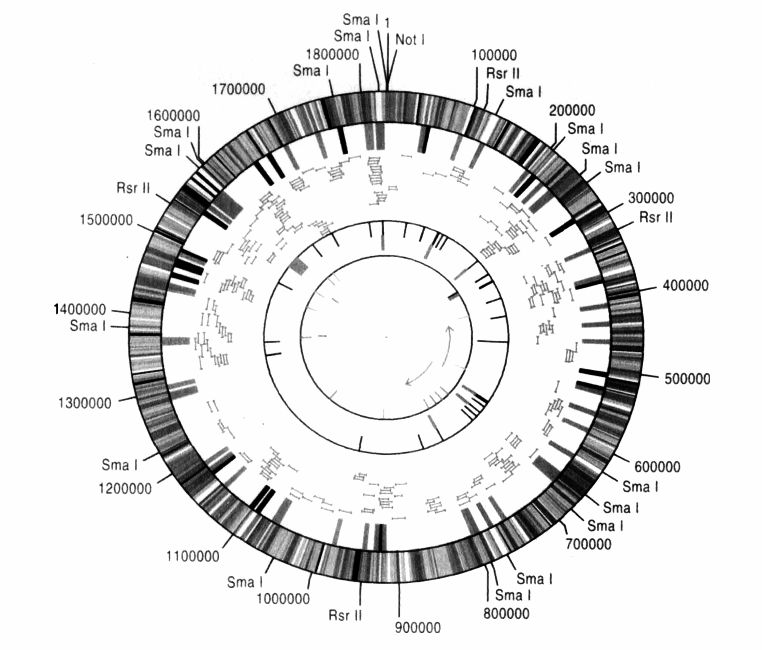
完整的基因组。流感嗜血杆菌的基因图谱:180万个碱基对,1727个基因。
我们是否能无中生有,用人工结合个别的纯化成分,制造出一个能够发挥生命功能的最小细胞?有鉴于还有超过100个生殖道支原体的蛋白质的功能仍然是谜,要达到上述目标,可能还有一段漫长的路要走。支原体有500种左右的蛋白质,其中有些蛋白质是由大量分子组成,有些只有一小撮分子,它们所构成的生命系统已经极度复杂。就我个人而言,光是要看懂只有四五个主角的《高斯福德庄园》就挺困难的;想到要了解活细胞里不同要素之间复杂的互动关系,就觉得这绝对是项艰巨的工作。因为活细胞绝不是一个简单利落的迷你机器,如同布雷纳所说的,它们就像“一个蛇窝,许多分子相互扭缠在一起”。但是温特仍然很有信心,认为人造细胞的时代即将来临,而且他已经找了一批生物伦理学家来谘商,看看是否要继续尝试。他们跟我一样,认为尝试以这种方式“创造生命”并没有道德上的问题。如果真的成就这个伟业,也只不过再度证实分子生物学界大多数人早已知道的事实:生命的本质就是复杂的化学作用,别无其他。在一个世纪前,这种“大发现”会成为头条新闻,但在今日,这已经没什么大不了。惟有相反的结论,即细胞的生命不只是基本成分及其化学作用而已,那才会在科学界激起波澜。
DNA分析改变了微生物学的面貌。在广泛应用DNA技术之前,辨识菌种的方法在分析能力上极为有限:你可以记录培养皿上菌落的形态,用微显镜观察个别细胞的形状,或使用相对来说很粗糙的生化分析法,例如依据细胞壁的特征,把菌种分类为“阴性”或“阳性”的革兰氏检测(Gram test)。有了DNA定序法后,微生物学家等于突然拥有了一个辨识指标,而且这个指标绝对是每个物种所特有的。即使那些由于自然生长情况难以模拟,而无法在实验室里养殖的物种,例如栖息在海洋深处的物种,现在只要能从海洋深处取得样本,同样可以用DNA分析法来研究他们。
如今,在佛瑞瑟的领导下,基因组研究所仍是细菌基因组研究的龙头。他们在很短的时间内,定序超过20种不同细菌的基因组,包括一种导致溃疡的螺旋杆菌(Helicobacter)、一种导致霍乱的弧菌(Vibrio)、一种导致脑膜炎的奈瑟氏球菌(Neisseria),以及一种会引发呼吸道疾病的披衣菌(Chlamydia)。他们最大的劲敌是桑格中心的研究小组。这个英国团队是由巴瑞尔(Bart Barrell)所领导,幸好他不在美国,否则以他有限的学历,绝对无法登上学术界的高峰。他没有博士学位,而是早在DNA定序法问世前,于高中毕业后直接担任桑格的助手,从此踏入科学界。在开始研究细菌前,巴瑞尔是因为自动化的先驱而扬名,他用数个ABI定序仪来破解有1400万个碱基对的面包酵母基因组,成功解开其中的40%,而这时欧洲其他的酵母菌定序小组仍执着于使用人工定序法。巴瑞尔的小组后来率先完成结核杆菌(Mycobacterium tuberculosis)的序列,这种杆菌会导致一度有“痨病”之称的可怕疾病——肺结核。

基因组研究所的领导人佛瑞瑟女士。
在高中时,佛瑞瑟“觉得自己被同学排斥,因为修这么多科学课程的女生并不酷”。在伦斯勒理工学院(Rensselaer Polytechnic Institute)念书时,她首次对微生物产生兴趣,于是申请进入医学院就读。后来她没有接受著名的耶鲁大学的入学许可,反而去了纽约州立大学布法罗分校,因为她的男友要搬去多伦多。这令耶鲁的主管百思不解:“小姐,我希望你知道自己在做什么。”但她跟男友的恋情并不长久;1981年,她嫁给了当时在布法罗担任助理教授的温特。她回忆说:“我们的蜜月旅行是去参加一场(科学)会议,还在那里写了一份经费申请书。”
运用DNA来分析微生物的做法,用在医学诊断上极为成功。要有效治疗感染,医师必须先辨识引起感染的微生物。传统的方法是利用被感染的组织来培养细菌,这过程慢得令人发疯,特别是在时间紧迫的情况下。在运用快速简单,也更加精确的DNA检测来辨识微生物后,医师可以迅速采取适当的治疗方法。这种技术在国家紧急情况中也派上用场:2001年秋天美国发生炭疽热下毒事件,基因组研究所的调查员找出第一位受害者身上炭疽热细菌的序列,取得了歹徒所用菌种的“基因指纹”,希望借此追查到细菌来源,进而逮住罪犯。
在我们对微生物的基因组更加了解后,一个惊人的模式开始出现。我们先前已经看到,脊椎动物的进化就像一个累进的经济体:通过逐渐增多的基因调节机制,同一个基因可以做的事愈来愈多。即使有新的基因出现,它们通常也只是既有曲目的变奏曲。相对地,细菌的进化是一趟激烈得多的改头换面之旅,这个令人眼花撩乱的过程偏好输入或产生全新的基因,而不是仅止于修改已经存在的基因。
事实上,重组技术之所以能问世,要归功于细菌纳入新DNA(通常是质体)片段的卓越能力。因此,微生物的进化会留存有过去惊人的基因输入活动的痕迹,实在不足为奇。大肠杆菌在我们的肠道(和培养皿)中通常很和善,但是通过基因输入,它们顿时成为变种杀手。有一种大肠杆菌菌株所制造的毒素,不时会引起食物中毒事件(1996—1997年曾造成苏格兰21人死亡)以及“杀人汉堡”事件(Killer Burgers,汉堡食材中含有杆菌毒素),这就是它们大量从其他物种“借用”基因造的祸。
遗传物质通常是在亲子中“垂直”传递(从祖先传给后代),因此从外面输入DNA称为“水平转移”(horizontal transfer)。把正常的大肠杆菌和致病菌株的基因组序列互相比较后,发现它们有共同的基因“骨干”,由此可以确认这两个菌株是属于相同的菌种,但致病菌株有许多由变异的DNA构成的“小岛”,这是它所特有的。整体而言,致病菌株缺乏正常菌株中的528个基因,但却多出1387个正常菌株没有的基因,数量可说相当惊人。这样的变异把大自然最无害的产品之一变成了杀手。
在其他的致病菌中,也可以找到类似的证据,显示曾发生过大量水平转移。就细菌来说,霍乱弧菌(Vibrio cholerae)是很异常的细菌,因为它有两个分离的染色体。较大的染色体(长度大约300万碱基对)似乎是这个微生物的原始配备,而细胞机能所需的大多数基因也在这里。较小的染色体(长度大约100万碱基对)就像一个镶嵌图画,由从其他物种输入的大大小小DNA片段所构成。
复杂的生物,尤其是人类这种大型生物,天生就会严密守护本身内在的生化机制:在大多数情况下,如果我们不摄取或不吸入某一物质,该物质就无法大幅地改变我们。因此长期下来,所有脊椎动物的生化作用通常没有多少变化。相对地,细菌暴露在异常化学环境中的情况多得多;一个菌落可能会发现自己突然陷入大量有毒化学药剂中,例如家用漂白水等消毒剂。难怪这些非常脆弱的生物会进化出各种各样的化学技能。事实上,细菌的进化是由化学创新所推动的,它们发明新酶(或修整旧酶)来做新的化学把戏。我们最近才开始解开其秘密的一群细菌,就是这种进化模式最惊人和最具教育意义的例子之一。这群细菌被称为“极端微生物”(extremophiks),因为它们偏好世上最不适合栖息的环境。
我们已经发现有些细菌是栖息在黄石公园的热泉里(例如一种超嗜热古细菌“激烈火球菌”[Pyrococcus furiosus]在滚沸的泉水中繁盛生长,在摄氏70度以下的温度就会冻死),或是在海底喷口的超热水域中生存(深海高压使喷口周遭的海水无法沸腾)。有些细菌可以在像浓硫酸那么酸的环境里生存,有些则存活在强碱性的环境中。嗜热嗜酸菌(Thermophila acidophilum)是名副其实的极端微生物,能够耐高温与强酸。在石油矿的岩石里也发现了一些菌种,它们可以把石油与其他有机物转化为细胞能量的来源,很像微型的高科技汽车。其中一个菌种栖息在距地表一英里或更深的岩石里,在有氧气存在时就会死亡,这种细菌有个相称的名字地狱杆菌(Bacillus infernos)。
生物学曾经有一个重要信条:生命作用的所有能量最终都来自太阳。但是近年发现的微生物之中,最惊人的或许就是那些推翻这个信念的微生物。支持这种信念的例子是,在沉积岩中发现的地狱杆菌和以石油为食的细菌,往回追溯,也都与有机物质有关:阳光在远古以前就开始照耀动植物,而它们的遗骸最后形成了今日的化石燃料(石油、煤等)。但是,现在我们发现了一类“无机自养生物”(lithoautotroph),这种微生物能从完全由火山爆发所创造的岩石中取得所需的养分。这些岩石没有包含有机物质的迹象,也不含阳光普照的史前时代所留下的任何能量。无机自养生物必须利用无机物来建造本身的有机分子,它们实际上是以岩石为食。
我们对微生物世界的不了解,最明显的证据莫过于很晚才发现的原核绿球藻(Prochlorococcus),它们的浮游细胞在大海上可以进行光合作用。在每一毫升的海水中,可能就有多达20万个原核绿球藻,因此它们大概是地球上数量最多的物种。它们当然也在海洋对全球食物链的贡献上占有极重要的比例。但我们却一直到1988年才知道它们的存在。
从我们周遭神奇的微生物世界,可以看出无数世纪以来自然选择所呈现的惊人力量。事实上,地球上的生命史主要是一出细菌的故事;比较复杂的生物,包括人类,都是很晚才登场的。35亿年前左右,生命最早以细菌的形式出现在地球上。第一批真核生物(其基因包在细胞核内)大约在8亿年后才出现,但其后的10亿年,它们一直维持着单细胞的形态。直到距今5亿年前才有一些突破发生,最终带来了蚯蚓、果蝇等生物,以及人类。细胞的优势可以从伊利诺伊大学的沃斯(Carl Woese)率先根据DNA重建的生命树看出:这棵生命树是一棵细菌树,在较晚长出的小枝上才有一些多细胞生物出现。沃斯刚提出构想时遭到生物学界的强烈反对,但现已广获采纳。不过,以DNA为基础来建立生命树,仍有一些令人难以接受的含义:例如它们显示动物和植物的关系不像先前所想的那么近,跟动物关系最近的生物反而是真菌——人类与洋菇有相同的进化根源!
人类基因组计划已经证明,达尔文的进化论比他自己敢于想像的还要正确。所有的生物都因为共同的传承而互有关系,而这也是分子相似性的来源。一个成功的进化上的“发明”(在自然选择上有利的一个或一组突变)会代代相传。随着生命树的逐渐多样化(原有谱系分开,产生新谱系,例如爬虫类现在仍继续存在,但也衍生出鸟类和哺乳类的谱系),进化的“发明”最终可能出现在大量后代物种上。例如我们在酵母里发现的蛋白质,约有46%也见于人类身上。酵母(真菌类)谱系和最终造成人类出现的谱系,可能是在10亿年前分开的,后来这两个谱系独立发展,各走各的进化道路。事实上,从酵母与人类的共同祖先开始,进化就足足进行了10亿年;但是在这段期间,存在于共同祖先里的那一组蛋白质只有些微改变。一旦进化解决了一个特殊问题(例如设计出一种酶来催化特定的生化反应),就会一直沿用相同的解决方法。我们先前已经看到,这种进化惰性是造成RNA主导细胞作用的原因:生命始于RNA世界,而这个遗产一直存留至今。这样的惰性也延伸至更细腻的生化层次:蠕虫蛋白质中有43%跟人类蛋白质的序列相似,果蝇蛋白质有61%,河豚蛋白质则有75%。
比较不同生物的基因组也可以看出蛋白质的进化过程。蛋白质分子一般可以视为不同结构域(domain)的集合体,所谓结构域是具有特定功能或形成特定三维结构的氨基酸链段,而进化似乎是借由置换结构域,创造新的排列来运作的。大多数的新排列大概都是随机产生,不具功用,注定要在自然选择下消失。但是在一些罕见例子中,某个新排列证明是有利的,这时一种新蛋白质就会产生。从人类蛋白质中辨识出来的结构域,大约有90%也见于果蝇和蠕虫的蛋白质。因此,人类特有的蛋白质其实可能只是果蝇的一个蛋白质重新排列的结果。
生物体之间这种基本的生化相似性,最好的证明莫过于所谓的“拯救实验”(rescue experiment),这种实验是消除掉一个物种的某个特定蛋白质,然后利用从其他物种取得的相应蛋白质来“拯救”丧失掉的功能。这个策略已经用于胰岛素。由于人类与牛的胰岛素非常相似,因此无法自行制造胰岛素的糖尿病患者可以接受牛的胰岛素作为替代品。
下面这个例子很像二流科幻片的惊悚情节:研究人员借由操控控制眼睛生长部位的基因,让果蝇在脚上长出眼睛!这个基因会促使许多与制造一个完整眼睛有关的基因在指定的部位工作,让眼睛在那里长出来。老鼠体内控制眼睛生长部位的基因与果蝇的非常类似,所以在基因工程师的操控下,把老鼠的这个基因植入一个该基因被消除的果蝇体内,它便会发挥相同的功能。这样的事可谓相当惊人:套用先前人类与酵母同时沿不同路线进化的逻辑,果蝇和老鼠在进化上至少已经分开5亿年之久,因此这个基因其实已存续了10亿年以上。如果想到果蝇和老鼠的眼睛在结构与光学上截然不同,这样的事实更显惊人。每个谱系各自发展出适合其目的的眼睛,但决定眼睛位置的基因机制不需要改良,所以仍维持不变。
人类基因组计划最令我们感到谦卑的地方,在于我们发现自己对绝大多数人类基因的功能所知甚少。为了妥善运用得来不易的信息,我们必须设计出以基因组规模来研究基因功能的方法。
在人类基因组计划之后,“后基因组时代”有两个新领域出现,它们的英文名称都含有和基因组同样的字根-omic:蛋白质组学(proteomics)和转录组学(transcriptomics)。蛋白质组学研究基因编码的蛋白质;转录组学则致力于探索基因表现的位置与时间,亦即在一个特定的细胞里,哪些基因在转录上是活跃的。如果我们要了解基因组的实际动态表现,不应仅视之为一套组合生命的指令,而应视之为生命这出电影的脚本——一部按照精确顺序写下生命中所有应该上演的剧情的脚本,那么蛋白质组学和转录组学就是了解现场演出的关键。我们了解得愈多,看到的“生命电影”愈多。
我们很早就知道,从生物学观点来看,蛋白质绝不仅是一条线状的氨基酸链而已。这条链折叠成特殊的三维构造的方式,与其功能息息相关,而这也是蛋白质组学想要了解的。现在结构分析仍是依赖X光衍射:用X射线照射分子时,它们会在碰到原子后反弹并散开成一定的模式,从中可以推知三维形状。1962年,我以前在剑桥大学卡文迪什实验室的同事肯德鲁和佩鲁茨,分别以解开肌红素(将氧储存在肌肉里)和血红素(运送血液中的氧)的结构而得到诺贝尔化学奖。他们的研究具有划时代的意义。他们必须解读的X光衍射影像非常复杂,相较之下,DNA简单得多了!
有关蛋白质三维结构的知识,对医药化学家寻找新药帮助很大,许多药物就是靠抑制蛋白质的机能来治病的。药物研发日趋专业化与自动化,数家公司现在正试图决定蛋白质的结构,好像它们就是生产线上的商品。比起肯德鲁和佩鲁茨的时代,现在这类研究工作简单得多了:如今我们有更强大的X光源,自动化的数据记录,还有更聪明的软件让计算机速度变得更快,所以解开一个结构所需的时间已从数年减少至数星期。
从三维结构本身经常看不出蛋白质的功能,但是研究神秘的蛋白质与其他已知蛋白质的互动,可以提供重要线索。有一个简单的方法可以辨识这类的相互作用:在显微镜载玻片上放一组已知蛋白质的样本,然后把神秘蛋白质洒在它上面;这些神秘蛋白质事先已经过处理,在紫外线照射下会发出荧光。当神秘蛋白质“黏”到载玻片上蛋白质网格的某个位点,这表示它在这个位点上已和另一蛋白质接合在一起,这使后者也变得会发荧光。由此便可以推知,这两个蛋白质在细胞内会产生互动。
在理想状况下,要知道生命脚本、要看到生命电影,我们必须找出在个体发育期间的蛋白质组成过程中所有的精确变化,从受精时刻开始一直到成年时期为止。我们会发现,许多蛋白质在整个过程中都发挥作用,但有一些蛋白质在特定的发育阶段才有用,所以在不同的成长阶段我们会看到不同的蛋白质组,例如成人和胎儿的血红素便有略微不同。同样地,每种组织会制造其专属的蛋白质。
要找出一个组织样本中不同的蛋白质,仍是长久以来所使用的方法最可靠,亦即利用蛋白质分子的电荷和重量差异,以二维凝胶电泳法来分离它们。然后用质谱仪(mass spectrometer)分析分离出来的数千个蛋白质小点,定出每个蛋白质的氨基酸序列。不幸的是,要应用这类的蛋白质组学来分析由整个基因组编码的大量蛋白质,所需的经费往往超过学术界的科学家所能负担。因此,这类昂贵的研究大多是由经费比较充足的大制药厂研究人员来做。但是由于这个方法本身的限制,如果蛋白质的含量很低,即使是这些实验室也无法每次都成功。
由于这类高处理量的蛋白质组学需要昂贵的硬件设备,还必须要有产业规模的复杂自动化程序,因此今日大多数科学家在研究整个基因组的基因功能时,并不使用这种方法,大多釆取转录组学的方法,因为它比较便宜,也比较容易实施:一个基因组里所有基因的机能,可以借用测量个别信使RNA产物的相对数量来追踪。如果你对表现在人类肝细胞里的基因感兴趣,你可以分离出肝脏组织内信使RNA的样本。从这个样本可以大致了解肝细胞内的信使RNA族群。在信使RNA样本里,最常被转录和制造出许多信使RNA分子、非常活跃的基因,所占的复本比例会较高,而鲜少被转录的基因则只有少量复本存在。
转录组学的关键在于一个相当简单的发明,即DNA微阵列(DNA microarray)。你可以想像一个显微镜的载玻片,上面有蚀刻了3.5万个点状小井的格网;然后利用精确的微小取量(micropipetting)技术,在每个点状井放入一个基因的DNA序列,让格网里包含人类基因组的每一个基因。重要的是必须知道各个基因的DNA是位于载玻片上哪个位置。斯坦福附近的Affymetrix公司甚至已经进一步缩小这些数组,把它们蚀刻在小如计算机芯片的一块硅芯片上,这便是“DNA芯片”(DNA chip)。
使用标准的生化技术,你可以为肝脏信使RNA加上化学标签,例如让它们在紫外线照射下发出荧光,如同前述的蛋白质。接下来的步骤更凸显出微阵列法功能的强大及简单:你只要把信使RNA样本放到微阵列3.5万个装满基因的棋盘状小井里,使双螺旋体两股结合的碱基配对键,会驱使每个信使RNA分子跟原先产出它的基因配对。这种互补配对万无一失。来自基因X的信使RNA只会跟在微阵列上基因X所在的点结合。接下来只要观察哪些点与发荧光的信使RNA结合。如果微阵列上的某个点没有荧光,就代表这个样本里没有互补的信使RNA,因此我们可以推论这个基因在肝细胞里没有活跃的转录作用。反之,许多点发出了荧光,有些还特別亮,这显示许多信使RNA分子已经连结上去。结论是:这是个非常活跃的基因。因此,只要有一个简单的实验分析报告,就可以辨识出活跃于肝脏中的每一个基因。我们之所以能巡游分子世界,要归功于人类基因组计划的成功,以及它带给生物学家的新思维:我们不必再满足于研究零零碎碎的细节,现在我们可以纵览分子天下,一睹其最壮观的全貌。
难怪斯坦福的布朗(Pat Brown)视DNA微阵列为“一种新的显微镜”。布朗是应用这个方法的佼佼者之一,对这项技术揭露基因世界崭新全貌的潜力感到不可思议,他曾经表示:“我们现在就像才刚开始探索世界的学步小儿。”
转录组学不仅是一项卓越的技术创新而已,它也让我们在追踪致病基因时更上一层楼。我们可以利用微阵列技术,研究健康与生病组织在基因表现功能上的差异,从而找出特定疾病的化学基础。这个逻辑很简单:我们对正常组织与癌症组织进行微阵列基因表现分析,然后找出这两者之间的差异,亦即找出在一个组织有表现,但在另一组织无表现的基因。一旦确认运作失常的基因(例如在癌症组织中表现过度或表现不足的基因),我们就可以找出目标,用精确的分子疗法来攻击这个目标,而不是使用具有广泛毒性、会同时摧毁健康与生病细胞的放射线疗法和化学疗法。
我们也可以运用这种技术来区分相同疾病的不同形式。在这方面标准显微镜的帮助有限:对通过目镜来观察的病理学家来说,癌症看起来都差不多,但它们在分子层面的差异是极大的。例如淋巴瘤细胞有多种形式,但这些光靠视觉难以区分,即便用放大倍率最高的显微镜也很难区别,但是这些细胞在基因表现上的差异却很明显,这对发明最有效的疗法极为重要。在谈到早期认为特定组织的癌全都有相同根源的看法时,布朗指出:“这就像把胃痛想成只有一个原因。辨识同类病症细微的差异,让我们能更适切地治疗这些癌症。”
在冷泉港实验室,魏格勒(Michael Wigler)以不同的方式来使用微阵列法:他不是把RNA加到微阵列上,然后寻找基因表现,而是加入癌细胞的DNA,建立呈现在肿瘤里的基因多样性。许多癌症是染色体重新排列所引起的。例如当一个染色体的片段在无意间复制,导致为促进生长的蛋白质编码的基因数目过多时,就有可能造成癌症。有些癌症之所以会发生,是因为失去了能为抑制细胞生长的蛋白质编码的基因。医师可以运用魏格勒的方法,检查来自同一人的癌组织和健康组织的活组织切片。癌组织的DNA以红色染剂加上化学标签,正常组织的DNA则用绿色。然后把这两种样本的混合物,加入包含所有已知人类基因的DNA微阵列。如同标准微阵列实验里的信使RNA,这些加上标签的DNA分子的碱基对会和阵列中互补序列的碱基对键结。癌细胞扩增的基因会呈现红点(因为和那一点键结的红标分子比绿标分子多出许多),没有癌细胞的基因会呈现为绿点(因为没有红标分子和它键结)。这类实验使得目前已知会造成乳癌的基因名单大幅加长。
我们在对付一种人类疾病时,总会发现自己像在黑暗中摸索。如果我们对正常情况下的基因表现多了解一点,我们就能更快找到问题核心,知道究竟是哪里出了问题,以及要如何修正。如果对于从受精卵正常发育为健康成人期间的所有基因发挥功能的时间与位置,我们都能有动态层面的了解的话,就能以此为基准,了解每一种病痛——我们所需要的就是完整的人类“转录组”(transcriptome)。这是遗传学的下一个“圣杯”,也是下一个需要巨额经费的大型研究计划。就短期而言,一个比较可能实现,甚至比较重要的目标是取得老鼠的完整转录组。先做老鼠而非人类实验的优点,在于我们可以在胚胎发育期间进行观察,以及用实验技术来进行干涉。即使是从老鼠身上收集这类相关的重要数据,也需要投资大量的时间与金钱。此外,如同DNA定序实验所证明的,更好的做法是先花些时间去完成更简单的模型生物转录组,尽量精进我们的技术,然后再开始研究老鼠,人类当然得摆在更后面。
细胞分裂:一个细胞的染色体(蓝色)进行复制,先沿特殊的“纺锤体"(spindle,绿色)排列,然后才分配至每个子细胞。高科技影像技术协助呈现出令人惊异的染色体华尔兹舞曲,而这也正是生命本身能永久存续的基础。
以微阵列法研究酵母细胞周期中的基因表现后,我们发现,光是细胞分裂的分子动态就已经极度复杂。这当中牵涉到800多个基因,每个都在细胞周期的某个时间开始发挥作用。我们在这里再度看到,进化不愿修补没有坏的东西:一个生物过程一旦成功地进化,只要生命持续下去,这个过程就很可能继续使用相同的基本分子。就我们目前所知,在酵母细胞周期期间主导发育的基本分子,在人类细胞中也扮演类似的角色。
最终,研究这三个“组”(基因组、蛋白质组和转录组)的目标都在于针对生命的组成与运作,建立精细至个别分子层面的全貌。如同先前所见,即使是最简单的生命,也复杂得令人迷惑,而且尽管近10年来在这方面已有可观的进展,但我们仍要面对许多艰巨的挑战。就复杂的生物体而言,要想了解主导他们发育的分子作用——也就是在四个字母组成的密码串的操控下,由卵到成体的神奇之旅最好的途径还是从果蝇着手。
自从成为摩根的研究对象后,果蝇向来是遗传学研究的重心;在其后的年代,通过持续的创新实验,果蝇至今仍是基因的金矿。20世纪70年代晚期,在德国海德堡的欧洲分子生物学实验室(European Molecular Biology Laboratory),纽斯兰—渥荷德(Christiane “Janni” Nüsslein-Volhard)和魏斯豪斯(Eric Wieschaus)开始了一项抱负远大的果蝇计划。他们利用化学制品诱发突变,然后寻找果蝇后代在非常早期的胚胎阶段所发生的阻断现象。传统上,果蝇遗传学家的研究对象一直是影响成体的突变,如同摩根所发现会造成白眼(而非红眼)的突变。但是纽、魏两人不同,他们把注意力放在胚胎上,耗费眼力,花了多年时间用显微镜寻找难以捉摸的突变,这也让他们进入了无人研究过的新领域,收获相当惊人。他们分析发现了好几组规划果蝇幼虫基本发育计划的基因。
纽斯兰—渥荷德和魏斯豪斯的研究工作也传达出一个更普遍的讯息:遗传信息是按等级规划的。他们发现,有些突变种的改变范围广泛,但有些突变种的变化则较为有限;他们根据这一点正确地推论出,效果广泛的基因是在发育早期发挥作用,亦即它们是位于启闭阶级的顶端,相对地,效果有限的基因是在比较后期才发挥作用。他们发现了逐级传达式的转录因子:一些基因启动其他基因,而这些被启动的基因又启动其他基因,依次类推。事实上,这种逐级传达式的基因启闭机制正是建造复杂个体的关键。这就像负责建造砖块的基因可以独立制造出一排砖块;然而在伙伴的协调运作下,它可以建造出一面墙,最终盖好一整栋房子。
一个个体若要正常发育,其细胞就必须“知道”自己在个体内的位置。毕竟,果蝇翅膀尖端的细胞所走的发育路线,应该跟果蝇脑部的细胞极为不同。第一条有关发育部位的必要信息最简单:发育中的果蝇胚胎怎么知道哪一端在哪里?头应该往哪边长?bicoid蛋白质是由母体的一个基因所制造的,它会以不同的浓度分布在整个胚胎中。这个效应称为“浓度梯度”(concentration gradient):这种蛋白质的含量在头端最高,愈接近尾端含量愈低。因此,bicoid浓度梯度会告诉胚胎中的所有细胞,它们在头-尾轴上的位置。果蝇发育是环节式的,它的身体分为数节,这些体节都有许多共同点,但每一节又各有独特之处。在许多方面,头部的组织方式跟胸部(昆虫身体的中间一节)相同,但头部有其特有的器官,例如眼睛,胸部也有其特有的器官,例如脚。纽斯兰—渥荷德和魏斯豪斯发现,有数群基因负责指定不同体节的身份,例如“成对规则基因”(pair-rule gene)为相隔体节的转录因子(基因开关)编码,所以“成对规则基因”发生突变时,会造成胚胎每隔一个体节就发生缺陷。
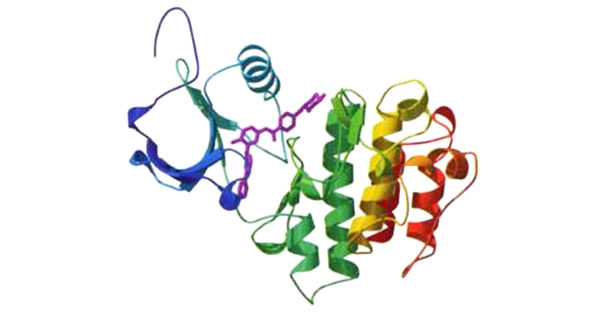
蛋白质组学:上图为致癌蛋白质BCR-ABL的三维结构。染色体异常使BCR和ABL这两个基因结合在一起,产生了这种蛋白质,它会刺激细胞增殖,而且可能引起一种形式的白血病。紫色部分是一种小分子药物Gleevec,可以抑制BCR-ABL的功能(见第五章)。在未来,这类三维结构的信息可以用于设计针对特定蛋白质的药物。这个BCR-ABL的结构模型没有显示出原子或个别氨基酸的细节,但仍精确呈现出蛋白质的整体结构。
1995年,纽斯兰—渥荷德和魏斯豪斯因为他们开创性的研究,荣获诺贝尔生理医学奖。他们在得奖后仍活跃于实验室,而不是退居以奖状装饰的宽敞办公室,这跟大多数桂冠得主不同。对魏斯豪斯而言,科学的魅力无法抵挡:“因为胚胎很美,也因为细胞的工作令人赞叹,所以我每天都怀着极大的热忱走进实验室。”他在亚拉巴马州的伯明翰长大,小时梦想当个艺术家。但他在圣母大学(University of Notre Dame)念大二时,由于钱不够用,就接下了科学界最臭、最卑微的工作之一:替实验室的果蝇族群调配“果蝇食物”(臭味冲鼻的胶状物,大多由糖蜜构成)。为数十万只肮脏又不知感激的昆虫下厨烹饪后,大多数的人可能会终生厌恶此种生物,但魏斯豪斯恰恰相反:他从此献身于研究果绳及它们的发育之谜。
纽斯兰—渥荷德出生于德国的艺术家庭,从小在自己感兴趣的领域样样出色,但对其他事物完全不理会。她在果蝇的发育遗传学上所获得的成果,足以让她发展两个职业生涯,但在获得诺贝尔奖后,她反而把她专注无比的注意力转移到斑马鱼(zebra fish)的发育上:这个新工作渴望解开许多脊椎动物的发育秘密。2001年,在诺贝尔奖的百年纪念活动上,我突然发现她是众多白发男士当中惟一的女科学家。事实上,截至当时在科学界只有10位女性荣获过诺贝尔奖,她是其中之一。
其他不再年轻的男科学家包括加州理工学院的刘易斯(Ed Lewis),他也是果蝇专家,和纽斯兰—渥荷德及魏斯豪斯共享这个荣耀。其实刘易斯不太像典型的银发族,虽然在参加斯德哥尔摩的颁奖典礼时,他已经年逾80,但在不必穿燕尾服时,他经常穿着慢跑装!他长期研究果蝇发育的基因调控,但对同源转化突变(homeotic mutations)特别感兴趣。这类突变会造成最奇特的结果:发育中的一个体节误拿到相邻体节的身份。在现在这个经常跟着流行制定科学计划的时代,一些价值观正逐渐消失,但是刘易斯在研究导致这些突变发生的霍克斯基因群(Hox genes)上所作的长期努力,却成了良好的典范。
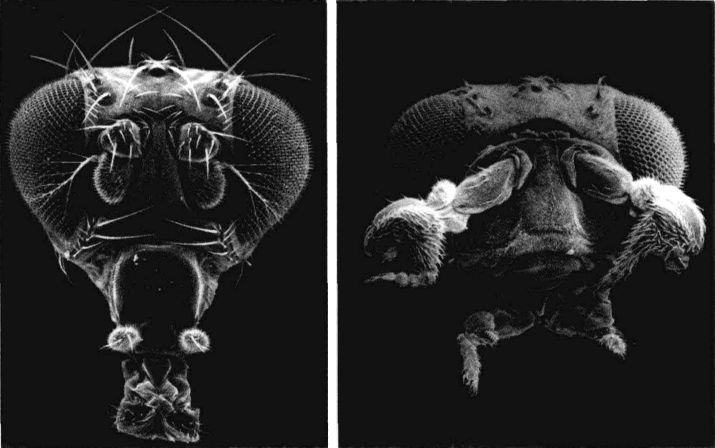
果蝇的脸:左图是正常个体,前额上长出一对有毛的触角。右图是触足突变种(antennapedia mutant),它的触角被完整的前足所取代。
现在我们已经知道同源转化突变会阻断为转录因子编码的基因(亦即基因开关),这种突变可能产生戏剧性的效果。触足(antennapedia)突变使果蝇在原本应长出触角的前额长出一对完整的脚。双胸型(bithomx)突变几乎同样怪异。在正常情况下,胸节之一会长出一对翅膀,而下一个比较接近尾端的胸节则会长出一对具有稳定作用的小结构,称为平衡棒(halter)。但是在双胸型果蝇突变种身上,原本应该长平衡棒的体节误长出翅膀,因此原本应该只有两个翅膀的果蝇变成有四个翅膀,而且第二对翅膀跟第一对同样完整。
正常运作的基因会调控体节的身份,确认每个体节的器官都长在正确位置上:头节长出触角,胸节长出翅膀和脚。但在发生同源转化突变时,各节的身份却发生混淆。以触足突变为例,头节以为自己是胸节,因此长出脚,而不是触角。值得注意的是,尽管脚的位置不对,但它的功能仍很完整。这暗示触足位置基因启动的是一整组的基因,它们通常是制造触角的基因或制造脚的基因(在异常发生时);但是在错误时间于错误位置被启动的整组基因,内部的协调工作并未受到阻挠。在这里我们再次看到,发育等级较高的基因控制了等级较低的许多基因。如同每个图书馆员都知道的,等级组织在储存和取得信息上效率较高。由于有这种等级式的安排,有时极少数的基因就足以造成很大的影响。
人类基因组计划缔造的惊人成就,引领我们进入了生物学最辽阔的一个新纪元。我们走在新研究领域“发育遗传学”(developmental genetics)的最前端时,竟然又折回去研究果蝇,好像有些奇怪。但我们必须“回到未来”,往回、往根源去探索,才能迈入未来,因为,即使已经解开整个人类基因组,但执行遗传指令的程序与线索仍是个巨大的谜。最终我们必会知道人类的生命脚本,如同我们研究的果蝇的脚本。我们将能完整地描述人类基因表现的模式(转录组),并且建立所有蛋白质作用的清单(蛋白质组)。我们将会对每个人极度复杂的组成全貌,以及构成我们的分子大军中的每一个是如何发挥功能的,有充分而完整的了解。