
南西·魏克斯勒(Nancy Wexler)在委内瑞拉马拉开波湖,抱着一位罹患早发性亨廷顿氏症的儿童。
在这时候就喝醉好像还嫌太早,更不用说还是一位穿着打扮完美无瑕的中年妇女,但是当她摇摇晃晃地过街时,看起来的确像是醉了,就连在附近法院执勤的警察也责骂她公开出丑。事实上,莉欧娜·魏克斯勒(Leonore Wexler)并没喝醉酒,她只是开始走向一个可怕的命运,一个在她眼前夺走了好几位亲人的命运,也是一个她原本希望能够幸免的命运。
不久之后,在1968年时,莉欧娜的前夫,精神分析医师米尔顿·魏克斯勒(Milton Wexler)原本要和他们的两个女儿——26岁的爱莉丝(Alice)和23岁的南西(Nancy)——一起在洛杉矶庆祝他的60大寿,结果那天的情况却变了调。米尔顿告诉两个女儿,她们的母亲患有亨廷顿氏舞蹈症(Huntington disease, HD),这种可怕的神经失调症会造成脑部功能发生愈来愈严重的退化,使患者逐渐忘掉自己与挚爱之间有关的事。他们也会无法控制自己的手脚,起初是走路受到影响,如同莉欧娜的例子,但是随着病情恶化,病人也会发生不由自主的痉挛。当时没有任何药物或疗法可以治疗这种逐渐步入死亡的可怕疾病。
现在爱莉丝和南西终于了解母亲的亲戚为什么会有那些令人不安的遭遇,她们也从母亲身上得知这个家族的问题。她们知道自己的三个舅舅,也就是莉欧娜的三个兄弟全都早逝;而且他们在死亡前,都有脸部歪扭、走路不稳、讲话模糊不清的情形。她们知道自己的外祖父,也就是莉欧娜的父亲沙宾(Abraham Sabin)也是年纪轻轻就过世了,不过莉欧娜很小心,从未提到她父亲在死前也有相同症状。这时爱莉丝和南西最终了解到自己的家族有亨廷顿氏舞蹈症。米尔顿必须承担的可怕任务是回答一个她们立即想到的问题:她们得这种病的几率是多少?他的回答是:50%。
令沙宾及其后代饱受折磨的亨廷顿氏舞蹈症,最早是由美国医师亨廷顿(George Huntington)确认的。亨廷顿出身医生世家,在长岛的东汉普敦(East Hampton)长大,从小就跟着父亲巡视病人。在哥伦比亚大学取得医师资格后,他回到长岛踵继父业,待了几年后才搬到俄亥俄州的波默罗依市(Pomeroy)。1872年,他在附近米德尔波特(Middleport)的米格和梅森医学院(Meigs and Mason Academy of Medicine)发表了一篇名为《论舞蹈症》(On Chorea)的论文。自17世纪起,医生就用源自希腊文中“舞蹈”之意的chorea来称呼会造成痉挛动作的病。后来亨廷顿在晚年时曾经详述他为什么会对这种神秘的慢性病产生兴趣:
50年前,有一次我和父亲去访问病人时,第一次看到“那种病”的病人,当地人总是用“那种病”来称呼这个可怕的疾病。那天的情景至今仍清晰得仿佛昨日之事,在我年轻的心灵留下永难磨灭的印象,也让我决定要以研究舞蹈症作为献身医学的处女作。当时我跟父亲正开车从东汉普敦前往阿默甘西特(Amagansett),在经过一片树林时,遇到两名背部佝偻、手脚扭曲、脸孔歪斜的妇人,我惊讶且几近恐惧地瞪着她们,心想,这是怎么回事?我父亲停车跟她们说了几句话,然后我们就继续前进。我由此展开了迦玛列式 的教育,我的医学教育也由此开端。从那一刻起,我对这种病的兴趣一直没有停止过。
的教育,我的医学教育也由此开端。从那一刻起,我对这种病的兴趣一直没有停止过。
这位年轻医生靠着本身的观察和父亲及祖父的临床笔记(祖父的原稿上有他父亲的铅笔注),在论文中精辟地描述了这种病症(后来即被称为“亨廷顿氏舞蹈症”,现今简称“亨廷顿氏症”。他指出,“当未受影响的肌肉也开始出现痉挛动作时”,舞蹈症的病况会“逐渐增加,直到所有肌肉都受到影响为止”。他也描述了伴随而生的精神退化现象:“随着病情发展,心智多少都会受创,最后有许多人发疯,有些人则是身心逐渐失常,直到在死亡中解脱为止。”他也认定这种病会遗传:“当父母之一或二者皆患病时,他们中的一位或一位以上的儿女必定也会罹患这种病。这种病不会隔代遗传,只要有一代跳过,下一代就不会再患病。”
亨廷顿正确地辨识出这种遗传疾病的主要特征。他发现男女都有可能得病,而且知道它会代代相传。只要父母中有一方得病,每一名子女遗传到这种病的几率都是50%。至于谁会患病则全凭运气,有些家庭是每一个人都患病,有些家庭完全没有人患病。如果一名子女没有从亲代遗传到这个异常基因,就不会把这种基因传给下一代。今天我们已经知道亨廷顿氏症是由基因突变引起的,由于这种基因不会表现在特定性別上(亦即并非性联遗传),因此我们可以推论这种异常基因不是位于X或Y性染色体上。我扪可以把这个基因的正常版本称为H,突变版本称为h。由于我们的体染色体(autosome,即不是性染色体的染色体;亦称常染色体)是成对的,所以我们也会有两个成对的亨廷顿氏基因。有两个正常基因(HH)的人可以预期不会患病,但是,拥有两个异常基因(hh),甚至只有一个异常基因(Hh)的人则会发病。我们把这种模式称为体染色体显性遗传(autosomal dominant inheritance)。
由于一个人得到一个异常基因的几率,比同时得到两个异常基因的几率大得多,因此大多数亨廷顿氏症患者都是Hh型。他们有可能把H或h传给子女,造成子女有50%患病几率,如同米尔顿给爱莉丝和南西的答案。
在1968年时,医界对这种病的认知仅止于下列事实:它是遗传性的,它会杀死脑部特定区域的神经细胞,造成不可逆的病程。米尔顿决定向这个使他全家陷于恐惧的疾病挑战:他成立了遗传疾病基金会(Hereditary Disease Foundation, HDF),募集资金,并且要求政府提拨更多经费来支持亨廷顿氏症的研究。他的女儿南西也投入这份工作,她在密歇根大学拿到心理学博士学位后(她的博士论文是研究可能患病者的心理状态),逐渐深入基金会的事务。20世纪70年代,在发现惟有深入了解这种病的遗传原理,才会有真正的进展时,南西开始朝成为遗传学家的路迈进。
在委内瑞拉的马拉开波湖(Lake Maracaibo)畔,当地的村落不仅贫穷,还因亨廷顿氏症的患病率特别高而饱受折磨。若要解开这种病的遗传秘密,这里可以说是最佳地点。1979年,南西开始在当地收集DNA样本,以及记录家族病史,目的在于建立当地所有患者的家族系谱。就一般遗传学家而言,这是一份辛苦的工作,而对身为亨廷顿氏症患者的女儿、在未来有可能发病的她来说,这份工作的意义远不仅止于此。她在陌生的环境中,看到很熟悉的景象:这里的人住在架高在湖水上、屋顶用铁皮盖成的小木屋里,但他们走路时像喝醉酒般摇摇晃晃,就跟她遭受病魔折磨的母亲一样。自从1979年初次造访马拉开波湖后,她每年都会回来这里继续研究。跟她合作的当地居民亲昵地称她“LaCatira”,意指她有金黄色的长发。如同她的委内瑞拉籍同事奈格瑞提(Americo Negrette,他是第一个就马拉开波湖的亨廷顿氏症患病率提出报告的科学家)所说,她把他们当成家人,每次跟他们打招呼时都“真诚自然,毫不做作,也不摆高姿态,眼里尽是亲切”。
但是亲切只能缓和亨廷顿氏症对这么多人的残害,南西远征之旅的最终目的在于找到造成这种疾病的基因。但是她要怎么利用在马拉开波湖建立的系谱来找出这个祸首?这当中的关键在于人类遗传学的进展。
若要找到亨廷顿氏基因,南西和其他对遗传疾病感兴趣的人知道,他们必须像半世纪以前摩根和他的学生对果蝇所做的一样,只不过把研究对象换成人。如同先前(在第一章中)所见,摩根让拥有不同性状组合(例如白眼[相对于红眼]和卷翅[相对于直翅])的亲代交配,比较这些遗传标志同时发生在混种子代的比率,然后利用这些数据,决定控制这些性状的基因在染色体上彼此相隔距离的远近。但是人类遗传学的进展之所以落后果蝇遗传学,主要有两大原因。第一,基于道德和实际理由,不可能对人类进行当时盛行的遗传分析实验:你不能叫两个你感兴趣的人类交配,然后在两星期后分析他们的后代。第二,就算人类可以任意交配,他们也仍然缺乏遗传标志。摩根能追踪因为个别基因发生特定的突变而在果蝇外观上造成的简单又明显的差异。但是在人类身上,这么容易遗传又能轻易分析的性状并不多;就连眼睛颜色也是由数个基因,而非单一基因所操控。此外,我们可以用X光照射果蝇或使用其他会造成突变的制剂来增加遗传变异的程度;但是,也幸好是,我们没有可用于人类的这类方法。一直要到重组DNA时代来临后,这两大难题才得以解决。
在DNA定序时代,我们不再需要看得到的遗传标志,例如果蝇的白眼;只要有序列本身的变异就已足够,而且我们只要分析数代的DNA,就可以沿家谱追踪这类的DNA标志(也就是通过一些遗传杂交来追踪)。在南西开始系谱研究的前一年,这场科学革命就已经展开。而且如同许多科学进展,这多少也跟运气有关。
犹他大学有个一年一度的“仪式”:每年都让一小群研究生跟他们的指导教授一起到沃萨奇山脉(Wasatch Mountain)的阿尔塔(Alta)滑雪胜地,参加密集的研习营(顺便也滑点雪)。这个研习营通常都会邀请其他学校的权威科学家参加,以批判性的眼光审视每位紧张的研究生所提出的数据。1978年,受邀参加的重要科学家包括麻省理工学院的波兹坦和斯坦福大学的戴维斯(Ron Davis)。
波兹坦给人的印象是“脑筋动得很快,说话也非常快,而且经常边想边说”。戴维斯则安静内敛。这两人尽管风格同异,那年4月在犹他州时,却心有灵犀一点通。他们在聆听犹他大学教授斯科尼克(Mark Skolnik)的研究生讨论在许多摩门教家族中追踪到的遗传疾病时,突然同时相互对望,眼中流露出相同的顿悟。尽管他们两人都是研究酵母的专家,但就在那一刻,他们领悟到找出人类基因位置的方法!他们发现可以利用尖端的重组DNA技术,把摩根研究果蝇的那一套遗传分析法,套用到人类身上。事实上,当时科学家已经用DNA标志来绘制一些其他物种的基因图谱,但波兹坦和戴维斯日后率先开展出把这个技术运用于人类之路。这个称为“连结分析”(linkage analysis)的技术,可以根据特定基因地标的已知位置,确定一个基因的位置。原理很简单:如果没有给你任何其他的信息,你很难在广大的美国地图上找到春田市;但是,如果我告诉你,春田市位于纽约和波士顿之间,有了这两个地标之后,你要找到春田市就简单得多了。连结分析就是把这个原理运用在基因上:它建立已知基因标志和未知基因之间的关联。这个方法用在研究果蝇基因上非常成功,但是如同先前所见,由于已知的人类基因标志不足,这个方法一直无法用在研究人类的疾病上,直到波兹坦和戴维斯发现分子生物学的进展已经解决了这个问题为止。
让他们俩眼睛为之一亮的DNA标志,即是我们在前一章中提到过的“限制酶切割片段长度多型性”(RFLP)。当某个个体内由特殊限制酶切断的DNA序列,在另一个体内发生变化,以至于无法再由相同的限制酶切断时,即成为RFLP。(如前几章所述,限制酶只能剪断特定的序列,例如EaoRl限制酶只有在遇到GAATTC时才会切断DNA。这个序列发生在基因组的特定位置,但在突变后,有些个体的这段序列可能发生变异,例如变成GAAGTC。限制酶只能切断没有改变的原序列,无法切断发生变化后的序列。)这些是在DNA序列里自然发生的差异,而且最常发生在垃圾DNA中,因此不具任何作用。事实上,在我们的基因组中散布着数百万个RFLP。在阿尔他研习营后数个月,波兹坦、戴维斯、斯科尼克,以及当时在马萨诸塞大学的怀特,开始一起研究RFLP的想法。1980年,他们共同发表了一篇划时代的论文,预示分子人类遗传学新时代的来临。他们规划出一个明确的计划,说明RFLP可能的运用方式。他们解决了数学上的问题,找出了需要多少个RFLP才能确保人类基因组的每一点至少都在一个RFLP标志邻近的合理范围内——原则上,这些条件可以帮我们绘出整个基因组图谱。这就像在北美地图上标出足够数量的美国城市,然后只须利用某个未标示地点跟这些已标示城市的远近信息,便能相当准确地找出这个未标示地点的位置。但是就基因图谱而言,何谓“合理”范围?波兹坦等人估计,只要有150个均匀散布在整个人类基因组中的RFLP就够了。这个方法最直接的优点在于提供了辨识致病基因的新策略。他们可以从已有数代子孙罹患某种疾病的家族中取得患病和未患病者的DNA样本,然后利用重组法——测试RFLP,寻找可以追踪这个家族患病轨迹的那些RFLP。
1979年,在这篇论文发表以前,怀特曾在冷泉港实验室的一次会议中提出了这些构想。他特別提到“那些比较正统的分子生物学家私下有很多抱怨和不满”。他听到许多人对这个方法能否奏效抱持怀疑态度;即使认为这方法可行的人,也对于运用它的最佳方式看法不一。后来在一场讨论如何运用RFLP连结分析来寻找亨廷顿氏症致病基因的会议上,大家开始公开提出不同的意见。
南西·魏克斯勒希望能立即以马拉开波湖的系谱来进行连结研究,但是波玆坦和怀特认为,要用RFLP连结分析来寻找亨廷顿氏症或任何其他疾病的基因,都为时过早。他们认为还需要做许多基础工作(例如先行找到和绘出基因标志的位置),才能够把这个技术用于这类特定的目标。后来,南西执意照自己的想法去做,于是双方朝不同的方向发展:遗传疾病基金会努力寻找亨廷顿氏症的基因,而波兹坦和怀特则致力于完成人类基因组的完整图谱。
波兹坦等人的目标是寻找每条染色体上的RFLP标志,而且要找到足够的数量,才能确保基因组的每一点至少都在一个RFLP附近。但他们很快就发现最初估计的数目150个,必须往上修正。但是学术界的实验室,例如怀特的实验室,并未因此受挫,仍然着手分离RFLP,而商业生物技术界也很快加入了行动。
1983年,经验丰富的分子生物家多尼斯-凯勒(Helen Donis-Keller,当时是波兹坦的妻子)在波士顿的合作研究公司(Collaborative Research, Inc.,以下简称“合研”)内成立了人类基因组部门。她的目标是制作整个人类基因组的RFLP连结图谱,上面有足够的标志可供找出任何染色体上的致病基因。四年后,合研公布研究成果,那篇论文有个很恰当的标题:《人类基因组的基因连结图谱》(A Genetic Linkage Map of the Human Genome)。这个图谱上标出了403个位置(比波兹坦最早的估计多出许多),而且计算结果显示,人类基因组有95%都位于某个标志的合理邻近范围内(或者说可合理连结至一个标志)。对基因图谱的制作而言,那是很重要的一天,但是到了1987年,研究人员之间再度出现分歧和竞争的情形。
其中一个原因在于合研公司毫不客气地采用大学实验室的数据,却不公布自己得到的数据,这种做法令学术界感到气愤。(在这方面,合研首开同时使用产学两界研究成果从而两面得利的先河,后来温特和其他想从基因组获利的人也很快地在定序竞赛中釆取了相同做法。)比方说,法国免疫学家多塞(Jean Dausset)的做法就不太一样。他在1980年赢得诺贝尔生理医学奖后,获得慷慨的赞助,得以用自己的策略来建立人类连结图谱。他知道如果全球所有的研究人员都釆用同一组标准系谱(取自同一家族的DNA)来做的话,这个工作的完成速度将会快得多。因此,多塞在巴黎成立了人类多型性研究中心(简称CEPH),收集最适合用于基因分析的系谱:有三代存活于世、可以取得样本的大家族。最后CEPH收集的资料来自61个家族,其中包括许多怀特研究的摩门教家庭、南西在马拉开波湖研究的家族,以及由约翰霍普金斯医学院的麦库锡克(Victor McKusick)所记录的安曼教派(Amish)家族。CEPH让研究人员得以自由地使用取自这些家族的DNA样本,惟一的条件是使用者必须把他们的分析结果交给CEPH,以便整合至这个全球性的数据库。合研公司就充分地运用了这个资源的好处。
然而,对合研公司1987年公布的图谱最严重的批评,在于它的标志分布得很不均匀。在第7号染色体(与合研的目标之一纤维囊泡症有关)上有63个标志,但第14号染色体上只有6个。在标志少的染色体上,这些标志之间的距离要比整个基因组上的平均值大得多。怀特对合研公司的做法感到特别不悦。他找到的标志超过470个,但是他向来在一条染色体上已找出足够密度的RFLP时才公布数据。他评论说,“我们的数据比他们多出许多,但我们永远不可能像他们那样公布成果,因为我们认为仍有重要的缺口必须填补。”他拒绝接受合研公司那套冠冕堂皇的说法。不过,无论那些说法是否冠冕堂皇,合研公司的图谱证明了制作整个基因组的图谱是可行的,而且本身也是很重要的进展。

绘制一种疾病基因的图谱:为了方便起见,这里只用两代和一些个体来作说明。如果这个分析的统计数据要够精确,则需要大量相关个体的资讯。
结论:后代的疾病X只与RFLPc连接,因此它的位置可以被定位在第6号染色体的这个区域上,但是如同前述,在1980年那篇有关RFLP的重要论文发表后,有些人看到的是另一条路(南西就是其中之一)。在建立完整图谱的工作加足马力后,麻省理工的豪斯曼(David Housman)也卯起劲来进行波兹坦认为现阶段不可能做到的任务:找出亨廷顿氏症基因的位置。他把这件很难办到的事,交给了刚在他实验室完成博士论文研究的古塞拉(Jim Gusella)。现在建立图谱的工作将进攻另一个阵线。
波兹坦最初之所以会有悲观的看法,原因即在于缺乏标志:理论上RFLP很可行,但实际收集它们的工作才刚起步而已。事实上,怀特、多尼斯-凯勒和其他人要研究多年,才有可能使已知标志的数目增加至数百个。古塞拉是在RFLP时代刚开始时就加入,这工作再适合他不过了。不过,到了1982年,他还只有12个标志,5个是自己找的,7个是别人提供的。在此同时,南西则回到马拉开波湖,尝试调整她的系谱,找出谁嫁给谁,有哪些子女,谁是谁的表兄之类的关系。当地的习俗有时会成为阻碍:有些姓名相当普遍,而且许多人有一个以上的名字。南西设法建立了一个家族的家谱,上面有1.7万个名字!她和她的同事会定期空出一整天的时间来收集血液样本,这些样本必须一起送往波士顿,免得马拉开波湖的热带气候加速DNA的降解。
至于古塞拉,他并没有等待马拉开波湖的样本。我记得他是在1982年10月冷泉港的一场会议上,提出了他最早期的数据。他从爱德华州一个饱受亨廷顿氏症折磨的小家族取得样本,在12个RFLP中,他只挑了5个来做测试,一一检视它们是否跟这个疾病有关,结果一个都没有。我有种大海捞针的感觉,他就像只捞出一些海水一样。惟有仔细分析所有的海水,也就是整个庞大的基因组,或是有非常好的运气,才有可能找到古塞拉想找的目标。因此当他最后在下结论说,“现在要找到亨廷顿氏症的基因只是迟早的问题而已”时,我不由地在心里响应,“是啊,只不过得‘非常迟’而已。”
但是命运会奖赏勇者。古塞拉回到实验室后,对更多的RFLP标志进行了测试,结果他震惊地发现,标号G8的第12个RFLP标志,似乎跟爱德华州那个家族的亨廷顿氏症有连结关系的迹象。因此他急切地等待马拉开波湖的样本,一收到它们就开始针对G8进行测试。当时的兴奋感几乎抵挡不住:G8的确与这种疾病连结。到了1983年的夏天,古塞拉终于突破万难,才试过12个RFLP就找到了跟亨廷顿氏症的连结。但这真的不是普通的运气而已:这是第一次在没有性别连结的协助,在对这种疾病的生化基础也没有任何了解的情况下,在染色体上找到了一种人类疾病的基因位置。这突然打开了一个崭新的科学远景,看来我们终于可以精密地分析自古以来折磨人类的所有遗传缺陷了。RFLP证明它们的确是有效的工具,而且在人类基因组中可处理的部分追踪到亨廷顿氏症后,应用强大的基因克隆技术来分离出这个基因,真的只是时间迟早的问题了。
亨廷顿氏症会对成年生活造成可怕的打击,但是,在幼年时期就发病的遗传疾病会让患者几乎没有生存的机会,因此更加令人畏惧。在诊断确认后,我们大多能以残酷的精确性预测病童的一生。假肥大型肌营养不良症(Duchenne muscular dystrophy, DMD)是一种进行性肌肉萎缩症,而且是一种性联遗传疾病:致病的突变发生在X性染色体的一个基因上。两条X性染色阵中的有一条有这种突变的妇女,通常会因另一条X性染色体上的等位基因是正常的而不会发病。妇女同时有两个不正常基因的几率很小,因为带有这种突变基因的男性几乎部无法活到可以传宗接代的年纪。然而,如果有突变基因的染色体传给儿子,则这个男孩一定会罹患DMD,因为他没有另一条X性染色体来提供正常的基因复本。等到这个男孩大约5岁大时,他的父母会发现他很难站起来或爬楼悌。等到大约10岁时,他得坐轮椅,最后他可能会在快20岁或20岁出头时死亡。DMD并不罕见,在男童的发生率为1/5000。
搜寻与人类疾病有关的基因,大多不是大型研究机构或有魄力的企业家一肩挑起,而是由遗传疾病基金会这类组织所主导,这类组织是那些对某种特殊遗传疾病有切身之痛的人成立的。由于攸关挚亲的福祉,这些组织然比较愿意支持有风险或创新的研究,尝试一般大学或生技公司所不敢涉足的领域。
长久以来,美国肌肉蒌缩症协会,以及欧洲的类似组织,一直都很支持以了解DMD基本生化知识为主要目标的实验研究。在20世纪70年代晚期,细胞遗传学家(他们用显微镜研究染色体)提供了第一条遗传线索。他们在一小群罹患DMD的女童身上发现,她们的一条X性染色体短臂上,标号Xp21的位置有异常现象。这有没有可能是DMD基因的位置?
不久之后,伦敦圣玛丽医学院的威廉森(Bob Williamson)率先运用以RFLP为主的方式,搜寻导致纤维囊泡症和DMD的基因。他的同事戴维斯(Kay Davies)导找X染色体上的RFLP,测试它们是否与DMD相连结。结果她成功了,决定性的关键就在于它们的位置:Xp21区,这跟先前依据DMD女病童奇特的X染色体所作的预测一样。
当这些搜寻基因的“基因猎人”努力分离与亨廷顿氏症和DMD有关的基因时,另一场较安静的革命也悄悄在临床遗传学家的办公室展开。从一开始,南西和豪斯曼就知道,可连接至致病基因的RFLP不仅可以用于找出这些基因的位置,也可以用于诊断某个家族的成员是否携带突变基因。它们甚至可以用于筛检未出生的胎儿。假设有一个DMD家族,其中至少一个男孩已经被诊断出有这种遗传疾病——这个男孩相当于“指标病例”(index case),亦即最先显露出这个家族有DMD突变存在的病例。他的母亲带有突变基因,也有另一个正常的等位基因。她的姐妹也可能携带有突变基因,因此她们所生的儿子都有患病的危险。现在假设这名母亲再度怀了一名男孩,则这第一个儿子有50%的几率会得病。但是在RFLP的协助下,她的医生可以告诉她,这个胎儿出生后的命运。
首先,研究人员必须先分析患病男童的X染色体,找出在这个家族中连结至DMD基因的RFLP。接着,从含有胎儿细胞的胎盘或羊水样本取得胎儿的DNA。如果这个胎儿的RFLP跟患病男童的RFLP相符,我们可以相当肯定这个未出生的胎儿未来也会患病。为什么只是“相当肯定”呢?本书的第一章曾提及,当卵细胞产生时,成对的染色体会发生重组,交换DNA:两条第1号染色体会互相交换,两条第2号染色体会互相交换,两条X染色体也会,诸如此类。如果这个交换刚好发生在X染色体上RFLP标志与DMD基因之间的区域,则连结正常基因的RFLP有可能会变成连结至突变的基因(DMD基因)。根据经验,对于最先连结至DMD的RFLP,这种情况的发生率大约是5%,所以根据RFLP所作的诊断只有95%左右的准确度。这5%的不准确度是重组作用所造成的不可避免的结果。因此,虽然这种诊断已经代表极大的进展,但是若要达到100%的准确度,仍然必须找到这个基因本身,而不能只靠跟它连结的标志。
最后分离出DMD基因的关键在于一位名叫布莱尔(Bruce Bryer)的男孩,他的X染色体的短臂少了一大截。由于缺少的部分实在太多,因此布莱尔除了罹患DMD以外,还有另外三种遗传疾病。1985年,哈佛医学院的昆克尔(Lou Kunkel)推论他可以用布莱尔的DNA,从正常男孩的DNA中“钓出”正常的基因。昆克尔利用重组法,把布莱尔的DNA从正常DNA中去除掉,留下其余的部分,也就是应该含有DMD基因的DNA。这种减去法并非百分之百有效,但已足以让昆克尔能够用与Xp21区有关的基因标志,找到想要的DNA片段。
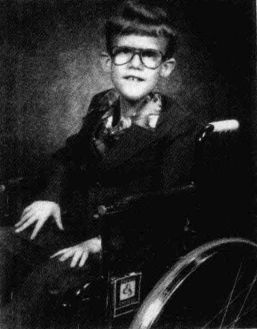
布莱尔的X染色体让科学家得以找出DMD基因。布莱尔是一位技巧高超的风琴弹奏家,他尽量设法过正常的生活,后来于一场车祸中丧生,享年17岁。
布莱尔的病例很特殊,因为他之所以得病,不是因为基因缺陷,而是因为他根本没有这个基因。昆克尔知道布莱尔所有的DNA在正常男孩的身上应该都找得到,而正常男孩拥有布莱尔没有的序列就是关键所在。
昆克尔指导的研究生摩纳柯(Tony Monaco)的任务是:如果Xp21区的DNA片段构成DMD基因的一部分,那么他得找出究竟是哪个片段。要做到这一点,惟有拿数个没有血缘关系的DMD患者的DNA,就每个片段——进行比对。摩纳柯在第八次尝试时终于成功:他发现有五个DMD男孩缺乏标号pERT87的序列。这意味着pERT87几乎可以确定非常接近这个基因,甚至有可能是它的一部分。摩纳柯开始分离pERT87附近的其他序列,结果证明DMD患者的DNA中也缺乏这些序列。到了1987年,昆克尔的研究小组已分离出整个DMD基因,现在它已经可以被定名为肌肉萎缩蛋白(dystrophin)了。即使在完成人类基因组序列后,肌肉萎缩蛋白仍是人类基因组中最大的基因,主要就是因为它有许多大的插入序列。
这个新知识很快就用于在产前确切诊断出DMD。科学家也很快就发现,有许多不同的突变会破坏肌肉萎缩蛋白,造成这种疾病。但当时他们仍不清楚这个基因的实际功能。了解它的功能是否能帮助我们发展出治疗DMD的有效疗法?
第一步是在肌细胞中找出这个基因所制造的蛋白质位置。在昆克尔的实验室工作的霍夫曼(Eric Hoffman)发现,肌肉萎缩蛋白一般位于肌细胞中,就在包覆肌纤维的那层膜下方。进一步的研究发现,肌肉萎缩蛋白在构成肌细胞内部结构的蛋白质,以及横跨细胞膜并与细胞外其他蛋白质互动的一组分子之间,扮演着重要的连接角色。细胞内部分子与细胞膜分子之间的连接,可以在肌肉收缩与放松时保护细胞膜。如果缺乏肌肉萎缩蛋白,细胞膜会受损,而肌肉细胞会一一死亡。我们对于肌肉萎缩蛋白已经有全新和详尽的了解,但至今却仍没有治疗DMD的方法,这可能很令人惊讶,但这也正是在目前的新科技中造成挫败感的主要原因:遗传学家已经能辨识和了解病因,但在大多数的例子中,我们仍然找不到矫正遗传错误的方法。
昆克尔的方法成为现代利用基因图谱来剖析疾病的典型做法。虽然现今这已是很常见的做法,但是在昆克尔的时代,这远远超出传统研究方法的范围,而美国肌肉萎缩症协会也是以孤注一掷的赌博心态,赞助了他的研究计划四年但他们的回收相当丰厚。先前我们只能利用生化方法来分析一个疾病的症状,试图找出这个疾病的基因;但是在昆克尔之后,我们可以制作这个基因的图谱,然后根据这个基因的功能来解释症状。
在支持图谱法的理由中,有一个最强而有力的论点是:这个方法在还没有完全辨识出疾病基因前就可以派上用场。在搜寻亨廷顿氏症或DMD的基因时所获得的基因标志,在这些基因本身被找出来之前就可以用于诊断。纤维囊泡症的情形也是一样。但是搜寻纤维囊泡症基因的研究之所以特别引人瞩目,原因有二:它是首度促使私人公司寻找人类致病基因图谱的疾病,它也是首度造成参与这类研究的科学家激烈竞争的疾病。
纤维囊泡症患者肺部会堆积浓稠的黏液,造成呼吸困难。肺气管壁细胞无法清除黏液,结果细菌在黏液中繁殖,造成肺部感染。在抗生素问世以前,这种疾病的患者只有10年寿命,但如今他们的存活率已改善很多。纤维囊泡症也是最常见的遗传疾病之一,在北欧血统的人当中,发生率约为1/2500。纤维囊泡症具有隐性遗传特性:一个人要遗传到两份突变基因才会发病。但是由于北欧裔中拥有一份突变基因(这些人本身有一份正常基因,所以不会发病)的比例多达1/25,所以两个各有一份突变基因的人结合,并把突变基因遗传给子女的几率相当高。因此,在研发这种疾病的诊断检测法成为实际可行的目标后,这立即成为医学界的优先工作。
在上海出生并在香港接受教育的徐立之(Lap-Chee Tsui),于1974年到美国读研究生。他在研究病毒时,获得有关分子遗传学的知识,1981年加入加拿大学者布赫瓦尔德(Manuel Buchwald)位于多伦多的实验室,研究纤维囊泡症。徐立之的个沉静、和蔼可亲,对研究目标非常专注,充满热情。他打算通过RFLP连结分析追踪纤维囊泡症的基因,于是把前两三年的光阴都花在寻找罹患纤维囊泡症的家族上,然后才开始用他拿得到的每个RFLP,对这些家族成员的DNA进行测试。但是他的运气没有寻找亨廷顿氏症基因的古塞拉好,在努力一年后,他所做的只是删掉了一大堆无关的RFLP。他需要更多的RFLP,而当合研公司愿意跟他分享他们的RFLP标志时,他忍不住欣喜若狂。
徐立之在多伦多的研究小组不是惟一致力于寻找纤维囊泡症基因的单位,伦敦的威廉森先前研究过DMD基因,现在也加入到寻找纤维囊泡症基因的行列,犹他州的怀特也在能拿到摩门教会所收集的大规模的家谱资料后,开始搜寻这种基因。摩门教的“祖先档案”记录,是为了让教友能够替生前不是教徒或在教会于1830年成立前就已过世的祖先祈福,好让家族永远团聚在神的国度中。宗教和遗传学的需求很少这么契合过。
后来,最先成功的是多伦多的研究小组。他们在1985年首度发现合研公司提供的RFLP之一和纤维囊泡症基因之间的连结。当时还不知道这个RFLP的位置,但是合研认为它具有金矿般的潜力,立刻展开搜寻。他们很快确认它位于第7号染色体上,但是没有立即通知跟他们合作的徐立之。他们在素负声望的《科学》杂志11月22日刊上公布这个发现时,也没有提及它位于哪个染色体。合研显然想独占这个新信息,但是在科学界是无法保持秘密的,有关它位于第7号染色体上的消息很快就流传开来。

基因追踪者徐立之。
合研公司保持沉默时,威廉森和怀特离有相同发现的日子也不远了。他们各自写了研究报告,交给《科学》的英国对手《自然》杂志,并且都提到这些关键的RFLP位于第7号染色体上。徐立之非常愤怒:在合作伙伴的欺瞒下,他即将失去宣称首位发现这个连结的权利(在科学界,亚军是没有任何奖赏的)。但是多尼斯—凯勒说服了《自然》也接受多伦多-合研研究小组宣布这个位置的论文。因此,最后在11月28出刊的《自然》杂志上,一共出现了三篇论文,编者还写了篇文章,解释三者的由来。
在这次学术与商业界的文化冲突后,多伦多与合研公同的伙伴关系就此破裂。后来合研发现,学术界跟他们合作时会十分提防,而合研执行长弗里德(Orrie Friedman)声称“我们拥有第7号染色体”,这种粗鲁而且不大站得住脚的说词对情况也毫无帮助。幸好这出肥皂剧终于在1985年12月落幕,所有的研究团队都同意分享自己的资源,以便针对211个家庭进行检测,确定第7号染色体上RFLP与病症的连结。结果相当惊人,这些RFLP跟纤维囊泡症基因非常接近,相距在100万碱基对以内,这使得这些RFLP在诊断上非常有用,而这正是纤维囊泡症研究的主要目标之一。
下一步甚至更加困难。知道纽约位于华府和波士顿之间,总比只知道它位于美国某处来得好。但是当你必须从华府徒步走到波士顿,一码码地寻找写着“欢迎来到纽约!”的标示牌时,光是知道这个线索恐怕也没多大用处。从连结分析的标准来看,100万个碱基对或许已经很接近,但是从基因克隆专家的标准来看,这可是迢迢长路,因为他们在分析DNA区段时,一次只做—个碱基对。为了找出最接近纤维囊泡症基因的两个RFLP之间的区段,徐立之和当时在密歇根大学的柯林斯合作(多年后,柯林斯继我之后成为人类基因组计划的领导人)。
当时柯林斯已经发展出“跳跃”技术,以便于克隆一对已知RFLP之间的基因,但是他跟徐立之一样,很清楚他们眼前的任务相当艰巨。在经过两年的研究之后,他们设法把纤维囊泡症基因缩小至28万碱基对的DNA区段内,并在这个区段内发现了已知于人类汗腺中扮演重要角色的基因序列,而纤维囊泡症患者正好有汗腺功能异常的症状。看来要找出完整的纤维囊泡症基因,指日可侍。
要确定他们找对基因的惟一方法是定出其互补DNA (cDNA)的序列,并寻找导致疾病的突变。以长达6500碱基对的区段而言,这在1989年是相当艰难的挑战,而且整个工作必须做两次:一次是使用纤维囊泡症患者的DNA,一次使用健康个体的DNA。最后的结果相当清楚:纤维囊泡症患者的DNA缺少由三个碱基对构成的小区段,造成这种蛋白质中少了一个氨基酸。大约有70%的纤维囊泡症患者都是这个突变所造成的,但是在纤维囊泡症基因中找到的其他突变也会造成这个疾病。由于有害的各种变异太多,使得以DNA为主的诊断工作变得格外复杂。现在我们把焦点转回南西·魏克斯勒、豪斯曼、古塞拉和他们的同事身上。先前曾提到,1983年他们成功地找出连结至亨廷顿氏症基因一个标号G8的RFLP。他们寻找亨廷顿氏症基因位置的速度快得惊人,看来他们大伙的运气异常地好,但是上天很快就把这种不平衡的状况矫正过来了。虽然他们只花了三年就找到这个基因,但是分离它以便进行深入分析的工作,却耗费了由150位科学家组成的国际研究小组十年的时间才完成。这个基因所在的区域长达400万碱基对。研究亨廷顿氏症的遗传学家辛苦地缩小着这个范围,但是随着基因距离愈来愈小,绘制基因图谱的工作也日益困难。最后他们的努力却只换来模糊的数据。这就像为了寻找纽约而从华府往波士顿走,却在走到费城的一个十字路口时,看到路标上有两个不同方向的箭头都指向纽约。
狩猎亨廷顿氏症基因的科学家决定改弦易辙,放弃矛盾的连结分析,把焦点放在亨廷顿氏症患者之间最相似的区段。这个方法终于将搜寻范围缩小至只剩50万碱基对,而这个研究领域也进入了基因克隆技术的时代。他们获得的第一批成果令人失望:在这个区段的右半边找到了三个基因,但是在亨廷顿氏症患者体内,这三个基因并没有任何异状。他们不屈不挠地又研究了这个区段的左半边,结果发现一个基因,并替它取了一个单调的名字IT15。在经过十年的努力,失败了许多次之后,命运之神终于再度对他们微笑。这个基因包含的一小段序列CAG,会像用于DNA指纹技术的STR般一再重复。最后他们发现,正常人的CAG重复数少于35个;CAG重复数超过40个的人,成年后会罹患亨廷顿氏症;在CAG重复数超过60个的罕见情况下,这人会在20岁之前就罹患严重的亨廷顿氏症。CAG是谷酰氨酸的基因密码,所以CAG每重复一次就会让这个蛋白质多增加一个谷酰氨酸。就亨廷顿氏症患者而言,由亨廷顿氏症基因编码的蛋白质含有额外的谷酰氨酸,这个差异可能对脑细胞里的蛋白质的行为造成影响,其原因可能在于它会使分子在脑细胞内聚合成黏性肿块,在不明机制下造成细胞死亡。
这是遗传疾病基金会研究团队的所有实验室辛苦努力的成果,而且为了表示这真的是协力合作的成就,在论文的作者位置只有“亨廷顿氏症联合研究团队”一个名称。当时已发现同样类型的奇特突变(三个碱基对的序列发生重复)也与另外三种疾病有关,令人惊奇的是,它们全都是神经方面的疾病。我们现在已经知道14种这类“三核苷酸重复序列型疾病”(trinucleotide repeat disorder),但是至今我们仍不清楚为什么脑细胞这么容易受这类型突变的影响。
听起来或许有些令人沮丧,但是尽管寻找造成亨廷顿氏症、杜馨氏肌肉萎缩症和纤维囊泡症这些疾病的基因耗费许多时间,但其实以遗传学家的标准来看,这些疾病都很“简单”。它们是由单一基因的突变所引起的,跟环境没什么关系。如果你的两份纤维囊泡症基因都缺少三个碱基对,或是一个亨廷顿氏症基因的CAG重复数超过40个,就会罹患这些疾病,跟你的居住地点和饮食都没有关系。目前已知有许多这种单基因病(single-gene disorder)——遗传疾病数据库已收录了数千种,但它们大多极为罕见,仅出现在极少数的家族里。
复合基因病(complex disorder),亦即多基因病(polygenic disorder)则普遍得多,其中包括许多最常见的疾病:哮喘、精神分裂症、忧郁症、先天性心脏病、高血压、糖尿病和癌症。这些疾病是由数个(或许是许多个)基因的相互作用所引起的,但这些基因的个别影响则很小,或许根本没有可察觉到的影响。一般而言,多基因病的问题更麻烦:这些互相作用的基因可能会造成容易罹患某种疾病的体质,但一个人是否真的会发病则取决于环境因素。假设你有一组基因变异,造成你有容易酗酒的体质,但你是否真的会酗酒在于你接触到的环境诱因,也就是酒。如果你在得州一个禁酒的郡长大,可能会跟在纽约市中心曼哈顿长大有截然不同的命运。同样的原则也适用于哮喘:即使在遗传上你有容易哮喘的体质,似是在“良好”的夏天,花粉和孢子的数量少时,你可能不会有任何症状。
基因和环境之间复杂的相互作用在癌症上最为明显。基本上,癌症是由数个基因发生突变所引起的遗传疾病。每个突变会改变细胞行为中的一个要素,直到这个细胞具有恶性细胞的全部特征。癌症突变的发生方式有两种。有些是遗传,我们都听过“那是家族遗传”的说法,有些被冠上这种头衔的特征不一定真的可以遗传(打个譬喻,就像天主教信仰),但是有些种类的癌症的确是有遗传性的。然而,由于癌症实在很常见,有时一个家族里有两个甚至多个与遗传无关的病例,也并非很罕见。(因此,研究“癌症家族”的遗传学家在决定一个癌症是否具有遗传性时,总是采取非常严格的标准。)有许多癌症突变也会在日常生活中发生。酶在复制或修补基因分子的过程中所犯的错误,或是细胞内正常化学反应的副作用,都有可能造成DNA遭到破坏。此外,许多癌症之所以发生,是因为我们自身的愚蠢。阳光中的紫外线是有效的致突变因子,而热爱阳光的人却心甘情愿地暴晒在阳光下;香烟能非常有效地把致癌物质直接送进人类的肺部,引起肺癌。现在已证明其他的环境因子,例如工作场所的石绵,也会助长癌症。这些例子告诉我们:DNA会自然受损,惟有靠着社会与个人作出明智的抉择,把这个损害降到最低。
1974年,以研究人类与黑猩猩的关系著称的玛丽-克莱尔·金转到加州大学旧金山分校,任职于研究乳癌的实验室,决心寻找乳癌基因。当时距RFLP连结法问世还有六年,但金已经知道家族系谱中藏有线索,于是她开始搜集家族。她寻找的是曾经有年轻乳癌患者及卵巢癌患者的家族,因为她推论这些病例可能是遗传造成的。当时她惟一可用的基因标志是蛋白质标志,几年后她发表了第一篇有关乳癌的论文,描述她在测试中并未找到乳癌与任何细胞表面蛋白质的连结关系,后来她发表的一些论文也获得类似的否定结果。反对者也同样秉持否定态度,他们认为乳癌受环境的影响太大,无法进行遗传分析,并预测这就像大海捞针一样。但是金愈挫愈勇,继续研究她的数据库,到了1988年时,在分析了1579个家庭后,她认为自己在这些高危家族中,找到了乳癌基因的好证据。
1990年,金在报告中指出,她研究了一个包括23个家庭的子群,在第17号染色体上找到一个连结至乳癌的RFLP,这个结果令医学界相当震惊。这些家庭在三代中一共有146个乳癌病例。金检视过可能干扰分析结果的因素,例如这些妇女可能接触过多的X光,或是她们的怀孕史跟其他人不同,但是她的数据仍站得住脚。在染色体上代号17q2l的位置有一个基因,当这个基因发生突变时,会使妇女罹病的几率大增。金的论文造成一股分离这个BRCA1(Breast Cancer之缩写,亦即“乳癌1”)基因的竞赛,也使基因的商业应用成为争议不断的话题。
分离BRCA1基因无疑是一件大事,即使它只对一小群高危家族具有重要意义(亦即它只是少部分乳癌病例的成因),然而光是与这个基因的功能有关的知识,就足以令人兴奋。金和柯林斯合作,柯林斯在寻找基因方面的资历无懈可击,但两人也遇到强大的竞争。先前参与RFLP连结研究并有重大成果的犹他大学族群遗传学家斯科尼克,跟吉尔伯特一起成立了万众基因公司。吉尔伯特担任Biogen执行长时历经的尴尬情况,并未消磨掉他的企业家精神。万众公司的计划是利用摩门教家庭的家谱来绘制和克隆基因,BRCA1很快就成为他们的目标之一。1994年,由万众公司、犹他大学、美国国家卫生研究院、麦基尔大学(McGill University)和礼来制药公司遗传学家联合组成的团队拔得头筹,宣布找到BRCA1基因“很有希望的候选者”,这说法算是相当客气,因为他们的确找到了正主。参与者全都提出专利申请(虽然万众公司起初刻意把国家卫生研究院的科学家排除在外)。1997年,万众公司的申请获得批淮。
在他们克隆BRCA1基因时,另一个由万众公司和英国癌症研究中心的遗传学家联合组成的团队宣布,他们在第13号人类染色体上找到第二个乳癌基因BRCA2。研究竞赛再度展开,短短一年内,英国团队就宣称成功分离出BRCA2。他们知道一旦找到这个基因2/3的DNA序列,而且证明6个不同家族的这个序列都有缺陷,就等于猎物落网。为了不落于人后,万众公司组成另外一个联合研究小组,这次是由加拿大和法国的机构所组成;他们很快就发表了完整的BRCA2序列,那是一个很长的基因。当然,万众公司和英国癌症研究中心都提出了专利申请。这些显然都是在商业上很重要的基因。这些基因的突变会对妇女造成非常严重的后果。妇女因为拥有一份突变的BRCA1或BRCA2基因,而在70岁之前罹患癌症的几率,有可能高达80%。此外,现在已经确定,同样的突变也会使罹患卵巢癌的几率提高,最高可达45%。对于家族中有突变病史的妇女,必须尽早让她们知道自己是否带有任何有缺陷的基因。现在已经有一些困难的但可能可以挽救性命的选择:高危妇女可以选择双侧乳房切除术(bilateral mastectomy),这可以使癌症发生率降低90%。同时,基因筛检可以找出这些家族中具有正常基因的成员,让她们安心地知道自己的罹病率不会特别高。
这听起来像是值得推行上市的产品:一种可以筛检严重疾病的基因检验,一个帮助妇女对自己的健康作出明智决定的方法。既然如此,为什么万众公司经常被指控为商业与科学结合做了最错误的示范?现在万众公司在美国拥有9项BRCA1和BRCA2的专利,2001年时,它又在欧盟获得1项专利,新西兰1项,加拿大4项,澳洲2项。事实上,这家公司垄断了这些基因,也控制着全世界使用它们的方式。万众公司从BRCA1和BRCA2的突变检验中获利是很合理的,毕竟他们提供了宝贵的服务,而且投资了许多金钱在研发这种检验上。但是这家公司要赚多少钱才算合理?今天这种检验的费用超过2700美元。同时,万众公司还限制学术界的研究人员使用BRCA基因序列来研发替代性的检验方法。对参与学术研究计划的病患进行DNA定序后,从中所得到与BRCA突变有关的信息也不得透露,就连病患本身都不能得知,否则就算是用在临床诊断上,也侵害了万众公司的专利权。
万众公司最近作了一些让步,他们和美国政府达成协议,凡是国家卫生研究院资助的研究计划,都能以1400美元的特价进行这项检验。但是批评者仍然认为这种做法徒具象征意义,而且批评声浪在加拿大和欧洲特别高亢。欧洲议会通过一项决议,表达对欧洲专利局的作为感到“沮丧”,并且责成议会的工作人员就万众公司对这两个基因的专利权提出抗诉。跟万众合作定序BRCA2的法国伙伴,对于万众的BRCA2专利感到格外愤怒,已向欧洲专利局提出联合控诉。无论如何,万众公司的垄断对病人恐怕都不是好事。这家公司的检验法并无法侦测出所有可能会影响基因的致癌变化,因此,筛检突变的捡测结果呈阴性的人,依然有患病的危险。现在当你在接受检测前,万众公司会要求你签一份弃权书,上面说明阴性的检验结果并不表示在遗传方面绝对没有问题。光是基于技术原因,要发展出更广泛的检验方法就已经很困难,但是如果没有万众公司打压研究的专利权,全球应该会有更多乳癌实验室从事这方面的研究。
过去10余年以来,科学家也把连结分析用于其他数种癌基因上,包括与纤维神经瘤病(俗称“象人病”,一般人并不知道这是一种癌症)、结肠直肠癌和前列腺癌有关的基因。这种一个基因一个基因来的方法尽管有效,过程却缓慢又辛苦,而且每一个研究都得找到适当的家族来进行分析才行。在这方面,人类基因组计划证明具有庞大的价值。本书第八章探讨的DNA与蛋白质微阵列法,提供给搜寻致癌基因的猎人们一种强大的绝佳武器。我在60年代刚对癌症研究产生兴趣时,我们对与癌症有关的遗传学所知甚少,而且只能依赖很原始的工具,以至于我只能转而研究造成动物罹患癌症的病毒,它们的基因数量很少,即使在那个年代也不难对付。当时我希望能借由研究这类病毒,对人类的癌症深入了解。今日癌症研究已不再限于病毒,我们已经有能力绘制人类肿瘤中数万个基因的图谱,并克隆这些基因。在我们发现所有使正常细胞变成癌细胞的细微生化异常情况,并对细节愈发了解后,将可以获得大量丰富的知识。
大多数的连结分析研究必须通过谱系来追踪基因目标,而且谱系愈大愈好。但还有一种策略是研究一小群某种疾病发病率特别高的人群,而要找到比特里斯坦-达库尼亚岛(Tristan da Cunha)还少的人群,恐怕很难。
1993年,“多伦多哮喘遗传学计划”检查岛上282名居民后,发现其中161人(占57%)有一些哮喘症状。加拿大研究人员建立了当地所有家庭的家谱;这个工作并不困难,因为所有的岛民都是早期15位开垦者的后代,因此他们的血统全都密切关联。哮喘显然是在1827年由两位妇女带入岛上的。对基因搜寻者来说,这样的人口用处极大:岛上居民就像一个大家族,所以引起任何可以察觉之病症的基因很可能在整个人口中都一样——这是最适合作连结分析的情况。在人数较多较为复杂的人口中,有些人的哮喘可能是一组基因所引起的,而其他一些人的哮喘则可能是另一组基因所引起的。这种异质性(heterogeneity)使我们很难找出造成复合基因病的决定因子。

从附近一座无人岛拍摄的特里斯坦-达库尼亚岛:这可能是地球上有人居住的最偏远地点。 特里斯坦-达库尼亚岛陡峭地矗立在南大西洋上,土地面积只有40平方英里。岛上首度有人定居始于1816年,英国派军驻守该岛,以防法国人以该小岛为基地,把遭到放逐的拿破仑从北方1200英里处的圣赫勒拿岛(St. Helena)救走。后来岛上的人口成长稀稀落落,有时来—些开垦者或几名船难幸存者。根据1993年非正式的人口调查,岛上只有301人。那一年,多伦多大学的一个小组来到岛上,就1961年对岛民所作的医学研究进行后续调查。在1961年时,岛上的休眠火山突然活跃起来,因此岛上的整个人口曾疏散至英国,在那里接受了健康检查。当时最惊人的发现是大约半数的撤离者都有哮喘病史。
多伦多的研究小组采集了血液样本并萃取出DNA,但需要许多经费才能完成研究。就在这时,他们和序析公司(Sequana)接触上,该公司的成立宗旨正是要找出疾病基因。序析公司赞助了这项研究,而这也立即引来这家公司剥削岛民的指控,岛民们对于自己在序析公司的商业策略中扮演的角色或许并没有充分的了解。自称为“促进农村发展国际基金会”的加拿大积极分子宣称,序析公司的行为“是一种生物剽窃(biopiracy)……违反DNA样本采集对象的基本人权”。序析公司宣称已经发现两个造成容易罹患哮喘体质的基因,但是在申请欧洲专利前一直拒绝公布它们,这种做法自然引起更多“生物剽窃”的指控。这些基因位于第11号染色体上,后续对高异质性的大陆人口的研究证实,第11号染色体的确与哮喘有关。因此,看来造成特里斯坦—达库尼亚岛上哮喘发病率高的遗传因子,不是只跟南大西洋海岛的孤立居民有关而已。数年后,史蒂芬森(Kari Stefansson)及其公司译码遗传(deCODE Genetics)也陷入类似指控的大风暴中,相较之下,序析公司历经的“生物剽窃”风暴可说是小巫见大巫。有鉴于要为每一种疾病都找一个类似特里斯坦-达库尼亚岛的小群人口来研究,是既冗长又没有效率的做法,史蒂芬森推论他需要的是一个人口大得多的孤立岛屿,这样可以一次在岛上居民中寻找数个疾病的基因。史蒂芬森刚好就是在这样的岛屿上出生的。
冰岛的面积跟美国的肯塔基州相当,但人口只有272512人,仅有肯塔基州的1/15。维京人于9世纪10世纪到冰岛定居,并在旅程中从爱尔兰绑架妇女过来。对于积极搜寻基因的猎人们来说,冰岛有数个优点。首先,冰岛人口的同构型高,几乎全部源自最早的定居者;从维京人的时代以来,外来的移民就非常少。其次,岛上保存了长达许多代、详尽的家谱记录;许多冰岛人可以追踪祖先至500年前。除了这个有用的资源以外,冰岛大学还保存了始于1840年详细的出生记录。最后,冰岛从1914年就开始实施全国性的保健,因此整个国家的医疗记录都经过整理,随时可以取用——至少原则上是如此。
史蒂芬森是哈佛大学的神经学家,对多发性硬化症和阿尔茨海默氏症(Alzheimer disease,即老年痴呆症)这类复合基因病很感兴趣。在认定自己的同胞最适合做遗传研究的对象后,他拟订了一个计划,连结谱系和医疗记录,建立用于搜寻基因的数据库。尽管这个计划的目的很有价值,但当地的隐私权法规却使这个计划窒碍难行,直到冰岛国会于1998年通过保健数据库法为止。这项立法授权“建立与管理无个人辨识数据的中央健康数据库,以增加知识,促进健康与保健服务”。
2000年,译码公司获得12年的授权,可自费建立与管理冰岛陕疗保健数据库,但必须缴年费给冰岛政府。这个数据库在家族谱系方面包含没有取用限制的信息,但医疗记录数据库则根据“假定同意”(presumed coment)的原则来管理,取用限制比较多。所谓“假定同意”是指除非民众主动选择退出,否则他们的医疗健康数据将会被输入这个数据库。在这个数据库里,对基因型数据的限制最严格,必须取得民众的“告知同意”(informed consent)才行,所谓“告知同意”意指个人必须主动同意提供组织样本以供取出DNA。这种做法成了争议的焦点:虽然译码公司采取保护捐赠者隐私的制度,批评者仍认为这是不够的。既然一个人的DNA数据必须连结至家族谱系和医疗记录,取样就不是匿名的。这些来源的身份是以加密方式来隐藏,而理论上密码有可能被破解。有关某个家族可能带有“不良”基因的小道消息可能会传开来,特别是在这种人口少的国家里,这种情形有可能导致“基因歧视”。译码公司的计划就像一个缩影,具体呈现出许多在其他地方还只是以假设性口吻来讨论的基因隐私权问题。
然而,尽管有这些争议,大多数的冰岛人仍支持译码公司,认为他们具有对抗遗传疾病的崇高宗旨,同时可望为冰岛这个小经济体带来大笔投资。在兴奋之余,许多冰岛人自己先大笔投资在这家公司,而在译码公司于纳斯达克正式挂牌之前,它的股票就已飙升至65美元。但是经济衰退对整体生技业来说都有不利的影响,特别是译码公同。在本书撰写期间,它的股票只值2美元左右。许多冰岛人都后悔被当时那股“买!买!买!”的热潮冲昏头。然而不可否认地,译码公司和罗氏药厂以及默克公司(Merck)获利极丰的合作,还是为冰岛带来大笔资金。但是随着冰岛政府提供了两亿美元的贷款保证,再加上译码公司被迫大量裁员后,无情的现实是,译码公司可能无法为冰岛带来原先预期的经济繁荣。
但是译码公司真正的检验不在于反复无常的股票市场,而在于它创造的科学。不幸的是,在这方面,不公开或延后公开的商业做法使得实际的情况难以评估。我们通过媒体发布的新闻得知,译码公司正对46种疾病进行连结分析,包括哮喘、忧郁症、癌症、骨质疏松和高血压。它已经发现23种疾病的连结标志,而且已经分离出造成周边血液疾病、中风和精神分裂症的基因。但是由于他们发表在科学期刊上的细节极少,所以我们有时很难分辨到底是真正的科学成果抑或是宣传。但是译码公司还是证明了它的确有能力作出有用的贡献:在2002年6月,译码公司的研究人员发表了人类基因组的新图谱,比人类多型性研究中心(CEPH)的旧图谱要详细得多。此外,在发表各界期待已久的精神分裂症研究成果后,译码公司的科学及商业前景都露出了曙光。
无论译码公司的未来如何,它三管齐下,在范围明确的人口中,结合医学、家族谱系与遗传等三种记录的全面性做法,显然具有庞大的潜力。因此,在把整个国家的人口置于基因检视之下的做法上,译码公司可说吾道不孤。例如,芬兰有600万人口,大约有35种发病率显著的遗传疾病,其中有些很独特,有些只是比其他欧洲国家常见,因此芬兰人自然会引起不少遗传学家的兴趣。其他国家也加入基因数据库的潮流。在2002年4月,英国开始了“生物银行”(Biobank)计划,爱沙尼亚政府也正在推动一个类似的全国性计划。这类大规模的人口研究最终可以协助我们追踪基因,甚至是那些最难捉摸的基因。
人类对遗传学的探索有漫长的历史,从我们早期的祖先对特征的世代传递产生好奇心开始,一直至今。然而,在这历程中,科学基础其实都很薄弱。达文波特想借由寻找他所谓“低能”的遗传基础,来支持他的优生计划,这种意图几乎不能算是科学。若要衡量这个领域的发展有多慢,从以下事实即可见一斑:长久以来定义人类这个物种的一项基本遗传参数,根本是错误的;一直到1956年,也就是发现双螺旋三年后,人类染色体的数目才正确地定为46个,而不是自1935年以来众所公认的48个。但是自从RFLP连结研究开始后,在20年间,我们以惊人的速度获得大量知识,原本贫瘠的领域开始蓬勃发展。在完成人类基因组的定序工作后,我们可能很快会找到所有重大遗传疾病的基因。接下来的问题在于:我们要怎么运用它们?
迦玛列,圣保罗的著名拉比和导师(《使徒行传》22;3),信奉全方位的知识涉猎及日常体验。