
戴维·维特因遗传性免疫系统疾病而极容易发生感染,他从小在隔离的无菌世界“泡泡屋”长大,成为最早的“泡泡男孩”。
戴维·维特(David Vetter)自从出生之后就没有直接接触过另一个人类,因为他罹患了严重综合型免疫缺乏症(severe combined immunodeficiency disorder, SCID)。他的身体无法制造B细胞和T细胞,由于这两种细胞都是免疫系统对疾病产生反应的重要成分,所以他只要轻微感染就会生病。
戴维的双亲在他出生前就知道他可能罹患SCID,他们的第一个儿子就是死于这种病。这次维特夫妇和医生们都已有所准备,他们很早就决定,如果这个婴儿证明罹患SCID,他会在无菌的隔离环境里生活,直到治疗SCID的方法问世,以医学进展的速度来看,这应该不会太久。戴维在1971年9月以剖腹产的方式来到人世,而且立即被安置在一个无菌保育器里。任何人跟他接触时,都必须使用内建于这个小保育器里的乳胶手套。随着他逐渐长大,他也被移至愈来愈大的无菌环境,亦即塑料“泡泡”里,但是有一样东西一直没变:乳胶手套。它们是他触摸外界任何人或物体的惟一方式。
结果,原先大家期望的疗法最后证明是一场空。戴维仍以“泡泡”为家,而他也引起全国的注意。美国太空总署尝试帮助他,为他制造了一个活动型生物支持隔离系统(Mobile Biologistical Isolation System),基本上,这是一套让戴维可以自由地到“泡泡”外探险的太空装。但是,就算太空装也只是另一种形式的“泡泡”。
移植疗法的进步看似充满希望,1983年10月,就在戴维过了12岁生日的一个月后,他接受姐姐捐赠的骨髓,动了骨髓移植手术。不幸的是,后来发现姐姐的骨髓内含有一种病毒,在戴维毫无防卫能力的系统里引起了恶性淋巴瘤。1984年2月,他被迫离开“泡泡”,住进加护病房。他在不久后去世,但至少在最后一段日子里,他终于能够体会到人类温暖的抚触。
幸好SCID很罕见,但是儿童发生遗传疾病的几率却十分惊人。事实上,大约2%的新生儿天生有严重的遗传异常。根据估计,在儿童医院的住院病例中,1/10与基因有直接关系,而半数左右与基因也有间接关系。遗憾的是,戴维的例子显示出我们现有的知识对大多数遗传疾病力有未逮之处:我们知道哪里出了问题,也能诊断,但是在治疗方面,我们能做的相对很少,遑论治愈。
SCID在流行文化中的形象颇耐人寻味。20世纪70年代,这种怪病被拍成了一部赚人热泪的电视影影片《住在塑料泡泡中的男孩》(The Boy in the Plastic Bubble)。到了90年代,泡泡男孩(Bubble Boy)竟成为情境喜剧《欢乐单身派对》(Seinfeld)中的搞笑角色。2001年,迪斯尼发行了一部庸俗的电影,内容是一个被困在泡泡里的男孩一连串愚蠢的冒险行动,片中没有指明他得的是哪种病,但明眼人一看就知道。 科学在面对这类可怕的疾病时根深蒂固的无力感,多少解释了为何原先的苦情剧会演变成苦中作乐的喜剧。但是对患者及其家属来说,这种无力感只是令人更加难受。特别是会造成身体日益衰退且无法挽回的疾病,只要一被诊断出来,无异于宣判死刑。在疗法付诸阙如的情况下,有些人宁可不知道自己可怕的命运,特别是在看到挚爰的亲人遭受病魔的摧残后。以上一章看到的南西·魏克斯勒为例,她得亨廷顿氏症的几率是50%,而且这种病已经夺走她的母亲和舅舅。南西为了找出这种遗传疾病的元凶,在马拉开波湖和美国的遗传学实验室经年累月地辛勤工作。然而即便她卓绝的奋斗最后终于得以分离出这种病的基因,找出致命的突变,但是找到治疗法的希望依旧渺茫。尽管她已经尽了最大的努力,让诊断用的基因检测法得以问世,她本人仍然表示不会接受检测,至少在可行的疗法有一线曙光之前不会。她宁可活在不确定中,也不愿知道这场输赢几率各半的赌局结果:她有50%的几率得面对身心衰退的噩运,这位活力充沛的女斗士,有朝一日可能只剩下一具毫无生气的躯壳。
科学在面对这类可怕的疾病时根深蒂固的无力感,多少解释了为何原先的苦情剧会演变成苦中作乐的喜剧。但是对患者及其家属来说,这种无力感只是令人更加难受。特别是会造成身体日益衰退且无法挽回的疾病,只要一被诊断出来,无异于宣判死刑。在疗法付诸阙如的情况下,有些人宁可不知道自己可怕的命运,特别是在看到挚爰的亲人遭受病魔的摧残后。以上一章看到的南西·魏克斯勒为例,她得亨廷顿氏症的几率是50%,而且这种病已经夺走她的母亲和舅舅。南西为了找出这种遗传疾病的元凶,在马拉开波湖和美国的遗传学实验室经年累月地辛勤工作。然而即便她卓绝的奋斗最后终于得以分离出这种病的基因,找出致命的突变,但是找到治疗法的希望依旧渺茫。尽管她已经尽了最大的努力,让诊断用的基因检测法得以问世,她本人仍然表示不会接受检测,至少在可行的疗法有一线曙光之前不会。她宁可活在不确定中,也不愿知道这场输赢几率各半的赌局结果:她有50%的几率得面对身心衰退的噩运,这位活力充沛的女斗士,有朝一日可能只剩下一具毫无生气的躯壳。
有时照顾患者比本身患病更令人难以忍受。乔治亚州汉普顿市的凯萝·卡利(Carol Carr)一直照看着她丈夫霍伊特(Hoyt),霍伊特在30多岁时罹患亨廷顿氏症。他姐姐罗丝琳已经因这种病过世,他哥哥则在获知诊断结果后立即自杀。凯萝辞掉工作,成为霍伊特的全职护士,照顾了20年。在霍伊特被诊断出来前,他们已育有三子,而当他在1995年病逝时,凯萝已经开始照顾两个年长的儿子蓝帝和安迪,就跟照顾她丈夫一样,喂他们吃饭、替他们洗澡、给他们吃药、帮他们上厕所。小儿子詹姆士也很快显现症状,绝望的凯萝只好把两个大儿子送到私人疗养院。2002年6月8日,她在疗养院枪杀了他们俩。根据《纽约时报》的报道,詹姆士说,亨廷顿氏症早在他心碎的母亲扣下扳机之前,就已经杀死了他的两个哥哥。
并非所有的遗传疾病都会因为医学无能为力而成为悲剧,最好的实例或许要算是促使一些食品(特别是汽水等不含酒精的饮料)包装上出现细字警语“含苯丙氨酸”的疾病。苯丙氨酸是一种氨基酸,但是患有遗传疾病苯丙酮酸尿症(phenylketonuria, PKU)的人无法处理这种氨基酸。
这个故事始于1934年的挪威。一位年轻的母亲决心要找出她4岁和7岁的两个孩子究竟出了什么问题,他们出生时似乎十分正常,但是较大的孩子到了7岁还无法完全靠自己上厕所,会说的字也没几个,更不用说完整的句子。这个病例引起生化学家及医师佛林(Asbjφrn Fφlling)的注意。在进行一连串检验后,他发现一个他认为跟他们的情况有关的生化异常现象:他们的尿液中含有过多的苯丙氨酸。但他也发现他们并非孤立的个案,他在挪威各地22个家庭里找到的34个相同病例,使他察觉到自己在无意间发现了一种遗传疾病。
现在我们已知PKU是编码苯丙氨酸羟化酶(phenylalanine hydroxylase)的基因发生突变听引起的,这种酶负责将苯丙氨酸转变成另一种氨基酸,即酪氨酸(tyrosine)。PKU是一种罕见的疾病,在北美的罹患率大约为1万人中有1人,而且具有隐性遗传模式,孩子要从双亲遗传到两份突变基因(父一份,母一份)才会发病。病童的羟化酶无法发挥功能,造成苯丙氨酸在血液中累积,伤害脑部发育及导致严重的智力障碍。预防方法很简单:PKU儿童只要从出生起即以苯丙氨酸含量低的饮食为主,就可以正常成长,也就是尽量少摄取蛋白质,不喝含人工糖精的汽水,这些是苯丙氨酸的两大主要来源。仅是营养来源一项就足以造成脑部正常发育和严重残障的差别。从儿童一出生就检测其PKU状况,当然很重要。美国医师盖斯瑞(Robert Guthrie)发明了一种简单的检测方法,可以得知血液中苯丙氨酸的含量,他积极推广这种方法,直到它成为新生儿的标准检测项目为止。自1966年起,每个新生儿都由脚跟抽血采样,分析苯丙氨酸含量。每年有几百万名新生儿接受盖斯瑞血液检测法,不需要检验DNA任一碱基对,就可以筛检出这种遗传疾病。在这个检测计划推行前,美国大约有1/100智障儿是PKU所引起的,现在每年只有少数病例而已。
20世纪50年代,用显微镜来研究染色体的细胞遗传学(cytogenetics)兴起。把这方法用于诊断后,科学家很快就发现,染色体数目异常(通常是多一条或少一条)必定会引起严重的机能障碍。这些问题根源于基因数不平衡,正常状况应该是每个基因有两份。这类情况不像假肥大型肌营养不良症或纤维囊泡症会在家族里遗传,但仍旧属于遗传的范畴:它们是因为产生精子与卵子的细胞分裂过程发生意外,而自发形成的疾病。
在这类遗传疾病中,至今最广为人知的是以英国医师唐恩(John Langdon Down)之名命名的唐氏症候群(Down syndrome)。1866年,唐恩在担任智能障碍疗养院的医务总监时,首度描述了唐氏症的特征。他注意到在他的疗养院里,10%的病人长得很像:“这情形实在太明显,在让他们并排站立时,很难相信他们不是手足。”然而一直到20世纪90年代后,法国医生勒热纳(Jérome Lejeune)才深入研究了这种疾病的生物学原理,他发现唐氏症儿童的某染色体有三条,后来发现是发生在第21号染色体。正常的情况是有成对的两条染色体,称为双染色体(disomy),所以唐氏症候群在遗传学上的说法是“三染色体21”(trisomy21)。
唐氏症的发生率会随母亲年龄的增加而升高。20岁的母亲产下唐氏症宝宝的几率大约是1/1700,但是35岁的母亲产下唐氏症宝宝的几率升至1/400,45岁的母亲则升至1/30。基于这个原因,许多高龄产妇选择对胎儿作产前诊断,判定其第21号染色体是否有三条。这个检测首次执行是在1968年,今日它已成为超过35岁的孕妇需接受的例行检查。
由于发育中的胎儿必须够大,才能安全地从其身上取出组织样本,因此这类诊断不能在怀孕前期进行。一般而言,孕妇是在怀孕第15周到第18周时接受羊膜穿刺术(amniocentesis),这种检测法是取出一些羊水来进行化验(羊水中自然含有胎儿的细胞)。另一种可以在怀孕第10周就进行的检测法,则是收集绒毛膜绒毛(chorionic villus)的细胞,绒毛膜绒毛是胎盘连结子宫壁的部分,但这种方法比较不可靠。由于两种方法都略具危险性(羊膜穿刺术造成流产的几率是1/100,而绒毛膜绒毛采样术造成流产的几率是2/100),因此一般不建议比较年轻的孕妇做这些检测。事实上,她们的胎儿有遗传缺陷的几率比这些检查术伤害胎儿的几率还低。取出的胎儿细胞以前一度必须在培养皿中培养,然后才能进行染色体分析。现在已经有了荧光原位杂交技术(fluorescence in situ hybridization, FISH),诊断速度更快。这种方法是把一个小荧光分子贴到第21号染色体特有的一段DNA序列上,再注入样本中,接着它会连结至胎儿第21号染色体的DNA。如果细胞核上有两个荧光点,这个胎儿是正常的;如果有三个荧光点,这个胎儿则有唐氏症。
在英国,30%的唐氏症胎儿是在对5%最年长的孕妇做例行检查时发现的。从平均每英镑支出的检测成果来看,这个方法显然很有效率(自从当时的首相撒切尔夫人抨击保健开支后,英国国家保健局经常采取这种计算法),但其余70%的唐氏症病例呢?年轻妈妈的胎儿的确比较少见唐氏症,但是这些妇女却占孕妇总人数的绝大部分。由于以统计上的几率而言,不值得冒险做这些标准检查,所以医界一直想寻找非侵入性的替代指标。最后发现母亲血液中有些可侦测的物质能够提供有用的信息。低α-胎儿蛋白(alpha-fetoprotein, AFP,即甲型胎儿蛋白)和高绒毛膜性腺激素(chorionic gonadotropin)与唐氏症之间有显著的相关性,但它们并不是三染色体的绝对指标。因此现代的做法是让比较年轻的妇女做血液筛检,如果显示胎儿有唐氏症的可能性,医生就会建议她们做羊膜穿刺术或绒毛膜绒毛采样法,以便进行确切的诊断。
遗憾的是,在今日,妇女得知自己的胎儿有唐氏症后仍然只有两个选择:成为唐氏症宝宝的母亲或流产。由于病情轻重程度不一,要作这种痛苦决定更加困难。唐氏症患者都具有唐恩发现的脸部特征(脸平而宽、鼻子小、眼皮窄而斜 ),但他们的智商差异很大,从20到85不等(亦即从重度迟缓到低度正常)。他们特别容易罹患多种疾病,包括心脏病(造成15%的唐氏症儿童在一岁内死亡)、肠胃道异常、白血病,而随着年龄渐增,也有可能罹患白内障和阿尔茨海默氏症,但是,他们也可能只有相对很少的健康问题。如今照顾的环境日益改善,而我们对多出的染色体所造成的风险也更加了解,唐氏症患者的预期寿命已经大幅增加,有50%的患者可以活到50多岁。尽管唐氏症患者在一生中,上医院会变得有如家常便饭,许多人会觉得这令人沮丧,但是一般而言,唐氏症患者仍可以享受人生,也可以为许多家庭带来欢乐。在有唐氏症患者的家庭中,父母承受的压力可能比较大,因为他们必须调整生活,照顾这个有特殊医疗需求的孩子,同时知道他们可能在许多方面永远不会长大。
),但他们的智商差异很大,从20到85不等(亦即从重度迟缓到低度正常)。他们特别容易罹患多种疾病,包括心脏病(造成15%的唐氏症儿童在一岁内死亡)、肠胃道异常、白血病,而随着年龄渐增,也有可能罹患白内障和阿尔茨海默氏症,但是,他们也可能只有相对很少的健康问题。如今照顾的环境日益改善,而我们对多出的染色体所造成的风险也更加了解,唐氏症患者的预期寿命已经大幅增加,有50%的患者可以活到50多岁。尽管唐氏症患者在一生中,上医院会变得有如家常便饭,许多人会觉得这令人沮丧,但是一般而言,唐氏症患者仍可以享受人生,也可以为许多家庭带来欢乐。在有唐氏症患者的家庭中,父母承受的压力可能比较大,因为他们必须调整生活,照顾这个有特殊医疗需求的孩子,同时知道他们可能在许多方面永远不会长大。

一名男唐氏症患者的染色体组型(karyotype),即全数染色体。从图上可以看出,他的第21号染色体多出一条,成了“三染色体21”。
得知胎儿有唐氏症的妇女通常都会选择终止怀孕。因此,在实施例行产前筛检的国家,唐氏症宝宝出生的数目日渐降低。然而在统计上,实情比这复杂:妇女晚生子女的趋势(经常是基于工作原因)其实增加了妇女怀唐氏症胎儿的几率。所以,在英国是以该年所有孕妇的年龄来预估可能会有的唐氏症宝宝数目,再以此为基准来衡量筛检计划的成效。现在我们看到的是唐氏症宝宝的比例不断下降;例如在1994年,筛检计划使唐氏症宝宝的发生率减少了40%左右。
三染色体的情况也会发生在其他的染色体之上,但是这些情况所造成的结果太过严重,因此孕妇都会自然流产,只有第13号和第18号染色体的三染色体情况例外。但是有三染色体13的婴儿大多只能活数周,而有三染色体18的儿童通常会在1岁前死亡。包括三染色体在内的染色体异常可能经常发生,许多染色体异常是会致命的,有些则没有或仅有极少的作用。据估计,目前总妊娠数中有多达30%是以自然流产结束,而在这当中,大约半数流掉的胎儿有某种形式的染色体畸变。染色体发生变化的后果可能远不如多出或少掉一整条染色体来得可怕,这些变化或许是一条染色体上有数个区段重新排列,或有一部分转移到另一条染色体上。如果这造成遗传物质净增加或净损失,如同多出或少掉一整条染色体的情况,这种不平衡的结果通常是有害的。不幸的是,对胎儿染色体进行一般的细胞分析,只能侦查到重大的失衡情况,但有时即使是较不重大的失衡情况也会造成非常严重的后果。
历经努力,在37岁第一次怀孕后,凯瑟琳·麦克欧利福(Kathleen McAuliffe)做了羊膜穿刺术,并放心地得知她的胎儿只有两条第21号染色体。但她不知道的是,从这个检测也可以看出其他的染色体异常。细胞遗传学家发现她胎儿的第2号染色体有倒位的情况:这就像这条染色体上有一个区段跑出来,翻了个身后,以跟原先相反的方向再嵌回原来的染色体上。但是除了这个消息以外,他们无法提供任何其他有用的建议,这个倒位有可能会造成问题,例如它可能造成遗传物质不平衡,但是它也可能完全没有作用。若要找到更多的信息,就得检查麦克欧利福夫妇的第2号染色体。如果他们当中任一人也有倒位情形(换句话说,这并不是胎儿的自发性变化),就可以推论这个倒位没有或只有极少的影响,毕竟双亲都是正常的。但是麦氏夫妇两人的第2号染色体都没有倒位现象,意味着这是在精子或卵子内初次发生的。这种倒位现象对胎儿有什么影响?凯瑟琳发现自己必须作一个攸关生死的决定。
痛苦许久后,凯瑟琳认为这种不确定性太大,决定终止怀孕。尽管她特别要求不要把解剖结果通知她,毕竟她仍为失去这个胎儿感到哀伤又充满罪恶感,但是在行政疏失下,解剖报告仍送达她家,而她发现胎儿真的有严重异常的情况。但这并不值得欣慰,至今她仍把胎儿的超音波影像收在抽屉里。幸好她后来再度怀孕,而且胎儿都没有任何问题。现在她是个幸福的妈妈,有两个小孩,而且如她所说,“他们健康得吵死人”。

用荧光染色来测定染色体数。一个细胞核(深蓝)的第10号(浅蓝)和第21号(粉红)染色体接受检测。左图的影像是每对染色体各有两条的正常染色体组型,右图是唐氏症染色体组型,其第21号染色体多了一条。
遗传知识会造成道德上的两难困境。凯瑟琳先前并未被告知羊膜穿刺术有可能查出三染色体21以外的情况,或许细胞遗传学家逾越了职责范围,他们应该仅针对当初做这个筛检的目的提出报告。当然,如果临床医生使用的是HSH法,就不会有这种问题,毕竟FISH法只能查出第21号染色体的数目。随着基因筛检法日益精进,就像打开潘多拉的宝盒一样,筛检结果的影响会远超过当初做这个筛检的目的,有时影响所及甚至不仅那些做筛检的人而已。在这方面,最明显的实例莫过于对有遗传病史的家族所做的基因筛检,例如有假肥大型肌营养不良症或纤维囊泡症的家族。在这些例子中,诊断不是由细胞遗传学家来做,而是分子生物学家,他们分析的不是一段段染色体,而是特定区段的DNA。他们先用羊膜穿刺术从胎儿身上取得组织样本,儿童或成人则从抽血或用压舌板从口腔内部刮得的细胞,取得组织样本,再从这些样本中萃取出DNA。现在这些检测通常会用PCR法扩增DNA样本的重要区段(可能有问题的基因),然后再作一系列的分析,以判定这个基因是否有突变。针对任何一人所做的检测结果,都可以告诉我们一些有关其亲属的基因状况。
有时这些影响不是发生在当下这一代身上,而是在未来的世代身上。X染色体脆弱症(fragile X)是遗传性心智迟缓最常见的形式。(唐氏症更常见,但为自发性,通常不是经由遗传得到。)除了智商低以外,X染色体脆弱症患者的典型症状是脸形特别长,下巴和耳朵特别大,极度好动,偶尔会有易怒的性情。这种病跟假肥大型肌营养不良症一样,都是性联遗传(导致这种病的基因位于X性染色体上),不同的是,男女都有可能得病。拥有这个基因的一个正常备份,显然不足以压抑突变基因的影响;但女性的症状通常比较轻微,而且女性的得病几率是1/8300,男性则是1/5000。造成X染色体脆弱症的突变,跟造成亨廷顿氏症的突变类似:它是因DNA三联体CGG一再重复而引起的。正常人的CGG重复数约为30个,但X染色体脆弱症患者的CGG重复数至少有50个,有时甚至多达90个。由于我们目前尚未完全明白的原因,CGG重复数一般会随着每个世代而增加;一旦有大约230个CGG三联体,这个基因将无法再制造信使RNA,从而无法再发挥功能。这种病的名称来由是因为这些重复会使X染色体的结构明显弱化,易于断裂。

6岁的唐氏症儿童JD和他的父亲。
以针对亨廷顿氏症所做的检测为例,在最近一个例子中,有一位20多岁的男士到提供遗传筛检(基因筛检)的诊所,要求做亨廷顿氏症的检测。他的祖父是因这个疾病过世,而他40多岁的父亲跟南西·魏克斯勒一样,选择生活在50%得病率的阴影下,不愿知道确切的结果。由于亨廷顿氏症是在人生中较晚的时期发病,因此很有可能这位父亲已带有突变,只不过还没有显现任何症状。这位年轻人知道自己带有这种突变,并在未来发病的几率是1/4,但他想知道确切的结果。问题是,如果他发现自己的确有这种突变,那么他必定是从父亲身上得到的,这代表他父亲未来一定会发病。这个儿子要求获知遗传信息的想法,跟他父亲不想得知真相的意愿发生冲突,家庭间起了纷争。最后是在这位年轻人的母亲介入下,他才没有进行筛检。她的论点是,她丈夫有权不想得知类似被判死刑的真相,相较之下,她儿子想得知真相的意愿自然较不重要。这个极端的例子凸显了基因诊断与其他诊断的不同之处。我从自己的基因得知的事,有可能对我的血亲造成影响,无论他们本身是不是想知道。
随着CGG重复数逐代增加,病情也愈发严重,而疾病征候的开始年龄也逐渐降低。X染色体脆弱症家族里最近一代的子孙,CGG重复数最多,而且通常发病年龄较早,病情也比他们的上一代严重。因此遗传学家可以找出携带“前突变”(premutation)的人,这些人的CGG重复数虽然尚未多到会在自己这一代引发疾病,但是从下一代的重复数有可能扩增的情况来看,已经足以使其后的世代罹患X染色体脆弱症。我们目前尚未确定这些突变基因所制造的蛋白质有哪些作用,但是它们似乎会结合至突触(synapses,神经细胞之间的接合区)中的信使RNA分子上。
如同亨廷顿氏症、假肥大型肌营养不良症和许多其他的遗传疾病,主导X染色体脆弱症相关研究的人,也是受这种疾病影响最深的人:患者的家人与挚爱的人。X染色体脆弱症协会(简称FRAXA)在募集捐款与促使国会支持X染色体脆弱症研究上,成效卓著。虽然有些科学家可能认为,这类团体只不过是提供那些陷入悲惨困境的人一些虚幻的安慰,让他们以为自己并非完全无能为力而已。经验显示,这些热心奉献、资源丰富,最重要的是动机强烈的组织,例如FRAXA,有时的确能突破万难,是破解这些疾病的重要关键。对于那些在财务和科学上都投下庞大赌注的人来说,有时在命运的协助下,会获得最大的报偿。
许多女性读到这里可能会产生一个问题:我怀孕时为什么没有做纤维囊泡症、X染色体脆弱症或假肥大型肌营养不良症的筛检?遗憾的是,这些妇女中有的人可能已有孩子罹患这类疾病。在使医学技术大转型的遗传学革命之后,有一个格外令人沮丧又没有道理的事实:科学进展与病人医护的脱节。事实上,比较正确的说法应该是,这点从未得到应有的重视,让两者立即挂上钩。无论如何,许多妇女根本就没有机会得知她们有哪些选择,而现在已经可用的检测方法,使用率也极低。
在主持人类基因组计划时,我必定会提拨经费,协助民众了解定序机器即将透露的知识会对众多人的生活造成什么影响,无论这些影响是好是坏。在提拨总预算的3%(后来是5%)用于这方面后,我任命亨廷顿氏症专家南西·魏克斯勒主持一个称为ELSI的小组委员会,专责探讨我们的研究在道德、法律及社会层面的影响。ELSI的主要计划之一是一系列有关遗传筛检的试验性研究。在每个新生儿都要接受苯丙酮酸尿症筛检的时代,我们的确有必要质疑:在面对纤维囊泡症、假肥大型肌营养不良症、X染色体脆弱症和其他科学可以预测的严重疾病时,医学连筛检这些疾病的选择都无法提供,是否是负责任的做法?当时是90年代早期,而今我们仍然停留在试验性阶段,除了一些零星的小规模研究,几乎毫无进展。造成这种停滞状态的原因有现实的钞票问题,也有人类生命的本质与尊严等深奥的哲学歧见。简而言之,这当中牵涉所有伴随遗传学革命而生的社会现象,从谋取经费到对生命的集体省思都有。目前有关假肥大型肌营养不良症和亨廷顿氏症的筛检,一般仅用于已经有一名成员发病的家族。这个限制的根据在于这些疾病很罕见,而这类筛检的费用很昂贵。这种社会成本计算法是有争论的余地,但相同的论据并不适用于纤维囊泡症的情况,尽管如此,纤维囊泡症的筛检仍有限制。先前提过,纤维囊泡症的发生率是1/2500,这使它成为最常见的遗传疾病之一,在北欧人之间尤为常见。有鉴于这种疾病的遗传缺陷是发生在第7号染色体,而且是必须同时有两份突变才会发病的隐性遗传模式,就令人觉得这种病的发生率实在高得惊人。只有一份突变的人不会发病,但会成为带因者,有可能把突变传给子女。根据流行病学的调查和估算,每25位欧洲血统的美国人当中,就有一位是带因者。
筛检纤维囊泡症的困难之一在于技术问题,与造成这种病的潜在缺陷的变异有关。在纤维囊泡症病例当中,大约有70%是由一种特定形式的突变所引起,称为△F508缺失(deletion),被删除的三碱基是CTT。如果其余30%只是由少数几种其他形式的突变所引起的,那么针对纤维囊泡症带因者进行全面性的族群筛检(population screening),便不至于不切实际。问题就在于大多数会造成纤维囊泡症的突变只发生在单一家族,而目前为止已发现超过1000种导致纤维囊泡症的不同突变。这在族群筛检上有何意义?实际上,任何筛检最多能找出25种不同的突变,但这25种最常见的形式仍仅占所有病例的85%。因此,大约每6个突变当中,就有1个不会筛检出来;以诊断来说,这样的命中率并不高。现在,假设有一对夫妇在接受极不完善的筛检后,两人都证明没有纤维囊泡症突变。但我们实在没法跟他们保证,他们的子女绝对不会得到这个病。人们难免要问:既然如此,那我们干嘛要花300美元做一个无法获得结论的筛检?
但是,尽管有这些技术上的困难,纤维囊泡症的产前筛检仍可以找出许多已经有这种病的胎儿。既然如此,为什么没有扩大实施筛检?矛盾的是,造成筛检范围仅限于有纤维囊泡症病史家族的主要推手,正是支持纤维囊泡症患者的团体。他们担心扩大筛检范围会瓜分有限的资源,使寻找疗法的终极目标获得的资源变得更少。这样的担忧是可以理解的,特别是现在。据估计,目前约有3万名美国人是纤维囊泡症患者。医疗上的进展已大幅延长了患者的预期寿命,而找到疗法应该也是可以期待之事。尽管如此,我们还是不能说疗法即将问世,这是不负责任的说法。患有纤维嚢泡症的婴儿,仍可能得和这种使身体日益耗弱的疾病奋斗一生。尽管治疗纤维囊泡症绝对是首要之务,但仍应让有意愿的孕妇有接受筛检的途径。然后她可以在对胎儿状态有充分了解的情况下,自由作出适当的选择。
有些人反对扩大筛检范围,是基于比较抽象的原因。有些人认为筛检等于自承失败,是一种错误的解决方式。支持团体的宗旨就是要让患者有社群归属感,觉得受到社会的重视。而筛检所促成的结果,直截了当地说,就是堕掉染病的胎儿,这不就和这些支持团体的宗旨相互冲突了吗?
支持纤维囊泡症患者的人一向尽量防止患者被打上某种歧视烙印,他们担心筛检会间接导致这个结果。事实上,在遗传筛检史上的确有过不幸的前例,令所有的病患支持团体难以忘怀。早在DNA筛检时代来临之前,最早期针对遗传疾病所作的诊断之一,是为了检测镰形细胞贫血症而发展出来的,在美国,罹患这种疾病的主要族群是非洲裔的美国人。如同第三章所见,那些同时拥有两个突变的“镰刀状”血红素基因的人,得承受身体逐渐耗弱的痛苦,而只有一个这种基因的人(亦即带因者)则几乎不会受到任何影响。
20世纪60年代,简单的血液检验发展出来以后,筛检计划在全美各地仓促展开。尽管立意良善,但它们造成的伤害却比带来的好处还多。筛检人员一般没有正确地告知检验对象有关这个筛检及其结果的重要性。许多诊断是带因者的人误以为自己患有这种疾病;有些人甚至因此找不到工作或无法参加健康保险。有可能把患病危险传给下一代的夫妇,被强力劝告必须考虑生孩子的后果。这些筛检计划(有些是强制性的)实质上形成一种压制,让一些人感到种族优生学又在美国复辟,检验结果呈阳性的人全都被打上了歧视的烙印。悲哀又讽刺的是,从纯粹医学观点来看,这个筛检运动其实是很有道理的:当时尽管在治疗上已有进展,但镰形细胞贫血症仍是痛苦的慢性病。对这种预防胜于治疗的疾病,筛检是最好的方法,但是最初设计来根绝它的机制在执行上却有很大的缺失,反而激怒了许多原本想造福的对象。
幸好在1972年,新的联邦政府施行方针重新设计了镰形细胞贫血症的筛检计划,让筛检能有效执行,但不会像第一次实施时造成人心惶惶。但是遗传疾病支持团体失去的信心仍很难弥补;这次经验让他们对筛检计划始终存有戒心,而对烙印的恐惧也挥之不去。可叹的是,有时这却损害到公共健康。
尽管遗传筛检在许多方面的确有用,但是总会引起争议。小儿科医师海格曼(Randi Hagerman)在丹佛儿童医院服务时,决定对丹佛特殊教育班的儿童进行X染色体脆弱症的DNA检测。她的理由很简单:如果能找出患有这种病的儿童,就可以给予这些因病导致学习能力受损的儿童更好的照顾,依照他们的特殊需求来规划课程。结果在439个受检学生中,发现5人有X染色体脆弱症。(在荷兰一个规模更大的学校调查,1531名学生当中,找到11个先前没有诊断出的X染色体脆弱症病例。)
在丹佛这项研究中,最有意思的部分或许是学生家长和监护人对海格曼提议的反应。他们大多认为这个诊断具有好处,可以改善孩子的教育,也能检验出家族血统中是否存有这种疾病。但是却有足足1/3的家长拒绝这项检测,理由不是说他们十分确定自己的孩子绝对没有X染色体脆弱症,就是担心这个检测会对孩子造成太大的压力。海格曼因这个计划饱受批评,反对人士群起而攻之。这些人坚信,试图以驾驭DNA来解决社会问题,无论如何,都会造成“基因极权政治”的危险。
这个问题不但攸关个人,确实也攸关社会。X染色体脆弱症的“前突变”发生率高(可能高达每200个X染色体中,就有1个有这种前突变),可以作为进行族群筛检的正当理由。在美国,根据估计,一个重症病人一生不工作并待在收容所里,大约要花掉200万美元。由于要提供负担得起的照顾愈来愈难,因此这一点成为提供每位母亲检测机会的有力论点。
这种基于严酷现实考虑的逻辑也适用于较小的国家,在这类国家,能够承受政策错误的资金更小。在以色列,一个对14334名妇女所作的试验性筛检研究发现,其中207人有前突变。在孕妇要求下所做的产前诊断,找出5个CGG重复情况已经扩增的胎儿。这些胎儿的命运当然是由孕妇来选择:自由社会不应要求妇女堕掉有遗传疾病的胎儿,也不应要求她将这样的胎儿生下。然而并非每位妇女都有扶养残障子女的准备,也不是每位妇女都会因为孩子未来的生活质量已可预见,而愿意终止怀孕。不过,无论个人的选择是什么,不变的事实是:筛检绝对会降低这种疾病的发生率,而这对社会毫无疑问是件好事。
尽管社会上存有抗拒心理,让大规模遗传筛检的好处无法发挥,令人沮丧,但在短暂的筛检史上,也并不完全都是小规模的实验性研究,以及一片挞伐之声。还是有一些快乐和发人深省的故事,说的是对高危族群进行遗传疾病筛检的成功案例。
血红素疾病(hemoglobinopathy)是由血红素分子的机能障碍所引起的疾病,包括多种地中海型贫血(thalassemias)和镰形细胞贫血症在内,一般认为它们是最常见的遗传疾病,全球人口大约有4.5%带有其中一种疾病的突变。先前已经谈到,镰形细胞基因具有抗疟疾的特质,所以在疟疾肆虐的地区会受到自然选择的青睐。因此,这种突变的高出现率最初仅见于世界上的这类地区。其他类似的血红素疾病分布模式,也是相同的适应优势所造成的。医学界早就知道,某些突变在一些民族中比较常见,无论属于这个民族的人身在何处。
在伦敦的希腊塞浦路斯裔族群中,地中海型贫血的带因者高达17%,相当惊人。严重的地中海型贫血是伤害力最大的血红素疾病,会造成畸形的、有时甚至会结核的红血球,引起肝脏和脾脏肿大,患者通常无法活到成年。1974年,皇家自由医学院(Royal Free Medical School)的摩戴尔(Bernadette Modell)展开一个有系统的筛检计划,结果这个计划受到伦敦塞普勒斯族群的热烈欢迎,因为他们太清楚这种病的严重性,长久以来他们的小区一直深受其害。意大利萨丁岛(Sardinia)1974年也开始进行类似的计划,使血红素疾病的发生率从1/250大幅减少到1/4000。
第九章提过的德系犹太人,也很清楚致命的突变会对在遗传上与他族隔离的族群造成什么后果。泰赛二氏症这种可怕的疾病在德系犹太族群中的发生率,比大多数的非犹太族群高100倍。罹患泰赛二氏病的婴儿在出生时看似正常,但后来他们的发育会逐渐变慢,日渐失明。到了2岁左右,他们会发作癫痫,情况持续恶化直到死亡,通常是在4岁之前,而且死亡时大多目盲并瘫痪。血红素疾病在特定族群中相对比较常见,通常可以用这是为了对抗疟疾而产生的适应保护来解释,但是泰赛二氏病不同,这种病在德系犹太族群中的高发病率,至今依旧是谜。遗传瓶颈或许是罪魁祸首:这种突变可能起先就存在于创始族群里相对较少的一群人身上,这群人在犹太人近代远离故土的第二次“大流散”期间分支出去,成为现今的德系犹太人。类似的现象或许也可以解释为何这种突变在魁北克西南部的法裔加拿大人,以及美国路易斯安那州的法裔卡真人(Cajun)当中格外常见:在他们所源出的一小群创始族群中,刚好有这种不幸的突变存在。另一个解释是:这个隐性基因(只有一份泰赛二氏症突变)的带因者对肺结核的抵抗力可能较强,这对历来倾向于以人口密集的都会区为家的欧洲犹太人来说,或许是一个优势。
1968年,在发现泰赛二氏症患者的红血球细胞中有过多的神经节糖RGM2(ganglioside GM2)后,泰赛二氏症的原因终于大白。神经节糖RGM2这种化学物质是细胞膜的基本成分,在正常人体内,多余的神经节糖RGM2会被一种重要的酶分解为相关的化合物,但是泰赛二氏症患者缺乏这种酶。1985年,梅洛威兹(Rachel Myerowitz)和她在美国国家卫生研究院的同事分离出了为这个酶编码的基因,证明泰赛二氏症患者的这个基因的确发生了突变。
从那时起,我们就可以根据这个基因,对明确的目标族群进行十拿九稳的产前检查,而这也正是最适合实施筛检计划的情况。但是在遇到阳性的诊断结果时,产前筛检惟一能提供的补救方法就是堕胎,而德系犹太族群——至少是那些严守教规的正统派——是禁止堕胎的。幸好,筛检也可以找出打算结婚生子的人是否身为带因者,所以对虔诚的教徒来说,符合道德观念的可行方法是以准备成婚的男女为筛检对象。纽约犹太教教士艾克斯坦(Yosef Eckstein)的10个孩子中,有4个死于泰赛二氏症。1985年,他设立了“正义世代”(Dor Yeshorim)计划,在当地的正统犹太教小区推动泰赛二氏症的筛检。他们鼓励年轻人在高中和大学时接受免费检测,这个计划的特殊之处在于高度保密:就连那些接受测试的人也不会得知自己是否是带因者,而是只拿到一个密码。未来等两个人打算结婚时,他们可以各自打电话给“正义世代”并提出密码。惟有在两人都是带因者的情况下,“正义世代”才会透露状况,并且提供咨询。这种有必要才告知的保密方式,目的在于避免让带因者被打上烙印,同时仍能对抗泰赛二氏症的威胁。
至今“正义世代”计划已经筛检超过7万人,发现超过100对有风险的情侣。这个计划使泰赛二氏病例不断减少,看来它是大大成功,但是仍有一些犹太人谴责这个计划。他们认为这个计划呼吁所有的年轻人接受筛检是一种压迫手段,同时认为它极力建议某些人重新考虑结婚决定的做法,是一种威吓。反对者把艾克斯坦的改革做法说成是“优生学”(对犹太族群而言,这是最让他们痛苦的一个字眼),但是这种具有煽动意味的说法根本无法动摇一个重要的事实:这个计划明显在它服务的小区获得热烈的支持,因为小区成员很清楚泰赛二氏症的可怕。事实上,艾克斯坦证明了筛检计划可以有效地进行,同时兼顾传统文化,甚至在社会习俗及宗教戒律与遗传筛检的原则看来难以兼容的情况下,也能行得通。
对于证实胎儿患有遗传疾病的孕妇而言,产前筛检提供了明确的选择:堕胎或继续怀孕。羊膜穿刺术只能用于至少已经15周大的胎儿,这个限制让选择堕胎更为痛苦。在这个阶段,堕胎所抹杀掉的不是一团没有特征的细胞,而是一个小生命——在超音波影像的协助下,胎儿显得非常真实,足以让父母对这个发育中的胎儿产生亲情。如果基于遗传筛检的结果而必须作出选择的话,大多数的父母——至少是那些并非坚决反对堕胎的人——绝对宁可在胎儿发育更早期的阶段作选择。这就是发明胚胎着床前遗传诊断术(preimplantation diagnosis)的灵感来源。

有8个细胞的胚胎。
在我们的文化中,人类的生殖生物学似乎是无穷争议的来源,而跟操控人类胚胎有关的做法,无论目的为何,势必成为攻击的焦点。胚胎着床前遗传诊断术也不例外,然而,撇开道德考虑不谈,这个程序仍有两大缺点:接受诊断术的夫妇必须有坚定的决心;此外,就跟所有形式的试管受精—样,这种技术非常昂贵。但基本上它的成效很好,造成的伤害也比堕胎少得多,现在只希望日后能有更好的技术问世,使成本跟着降低,这是科技发展的常见模式。在我们对抗遗传疾病的战争中,胚胎着床前遗传诊断术可能将是极重要的武器。

史蒂文森夫妇和子女合影:他们的长子特勒有X染色体脆弱症。胚胎着床前遗传诊断术确保他们的幼女莎曼莎没有这种疾病。
CNBC电视新闻记者黛比·史蒂文森(Debbie Stevenson)的儿子特勒(Taylor)患有X染色体脆弱症,但一直到她的第二个儿子詹姆士(James)出生后才诊断出来。尽管在50%的得病率,詹姆士却幸运地没有得病,但史蒂文森家仍不愿把第三个孩子的未来交由命运来决定。他们决定借助胚胎着床前遗传诊断术,黛比表示:“有些人认为选择健康的胚胎是不道德的,但我想这总比在得知你的宝宝有重大疾病时,才作是否要堕胎的痛苦决定来得好。”这个家庭在花了一整年的时间寻找到愿意执行这个技术的实验室后,黛比终于在2000年如愿通过这个技术怀了史蒂文森家最小的成员。在做过X染色体脆弱症的检査后,小宝宝萨曼莎(Samantha)证明跟詹姆士一样,没有遗传到特勒的可怕疾病。
伦敦汉默史密斯医院(Hammer smith Hospital)的温斯顿(Robert Winston)是一流的妇科显微手术医生,擅长于输卵管缺陷矫正手术(输卵管缺陷会使妇女无法受孕)。他也是在英国电视上推广科学与生物医学研究的健将,甚至有余暇以勋爵的身份担任上议院议员,就相关议题提供意见给政府。他结合试管受精(IVF)与以PCR为主的DNA诊断两种尖端技术,发展出一种方法,在胚胎殖入妇女的子宫并开始发育前,就检查这个胚胎的基因状态。数个胎体在试管里受精后,在实验室里发育,直到每个受精卵分裂三四次,有8到16个细胞为止。这时小心地从每个胚胎移出一两个细胞,取出DNA,再用PCR扩增相关的序列,以决定每个胚胎是否存有突变。PCR能扩增极微量的目标DNA,多亏了它这惊人的能力,这种能在胎儿发育极早期就进行诊断的方法才得以问世。然后父母便可以放心地殖入没有遗传疾病的胚眙。
于1989年首度施行的胚胎着床前检验,目的在于测知胚胎的性别;如果有可能罹患的是性联遗传疾病,例如假肥大型肌营养不良症,那么性别是相当重要的信息。一名是带因者的母亲可以选择只怀女胎,因为女孩虽然有可能是带因者,但是不会发病。后来把胚胎着床前遗传诊断术的用途,从简单的性别鉴定扩大至侦测特定突变的人,是温斯顿的同事汉迪塞德(Alan Handyside)等人;1992年,他们首度用这个技术来检测不是性联遗传的纤维囊泡症。
如同先前所见,尽管X染色体脆弱症是性联遗传,但男性和女性都有可能患病。因此,这种病自然会成为以特定基因为主的胚胎着床前遗传诊断术的目标,但是父母的态度仍要非常积极,而且明白扶养X染色体脆弱症的儿童非常辛苦,才能说服医生执行这个诊断术。
从遗传学的观点来看,目前为止我们讨论过的疾病都还算“简单”:它们都是由单一基因的突变所引起的,环境对一个人是否会罹患这些疾病毫无影响。但是癌症这类疾病则复杂得多,如同前述,癌症有可能在遗传与环境的合并影响下发生。但是即使是癌症,也有影响特别大的基因存在,无论环境的影响为何。虽然与乳癌有关的基因之一BRCA1,只占所有乳癌病例的5%,但是根据估计,这个基因发生突变的妇女到了60岁时,会有90%的几率得乳癌。
在20世纪90年代早期,当时任职于密歇根大学的柯林斯跟加州大学柏克莱分校的玛丽-克莱尔·金合作寻找BRCA1。他们采取标准做法:收集家族、准备DNA样本、测试标志,这一切都着眼于这个基因上。一个超过50人的家族中,有多起乳癌病例,显然这个家族的体质容易罹患乳癌。1992年9月,这个家族的一名成员(姑且称她为“安妮”)向柯林斯的同事韦伯(Barbara Weber)透露,她准备在下星期做双侧乳房切除术,虽然还没有迹象显示她已经罹患癌症,但她决定不再忍受对未来的不确定感,而想采取这种激烈的预防手段。然而韦伯从DNA分析中获得的结论是,安妮罹患乳癌的几率并不特别高,事实上,她得乳癌的几率跟没有家族病史的妇女一样。但是这个推论是以研究项目为背景的,而事前他们早已达成共识,这类数据不得用于临床诊断。
最后韦伯和柯林斯还是决定,安妮的困境比遵守规则重要。他们通知安妮,她得病的几率较低,于是安妮在松了一大口气后取消了手术。但是在把他们的发现告诉这个家族的成员之一后,这些研究人员觉得有义务让想知道的人享有相同的好处;于是韦伯和柯林斯特别成立了一个乳癌遗传咨询计划。这个家族中有一位证实患病几率并不特別高的成员,已经在五年前动了预防性双侧乳房切除术。她对这个迟来的诊断看得很开,她的想法是,动手术让她的心灵平静了五年。但是如果她的检测结果是有突变,那么当年这个激烈做法可以带给她的,可能不仅是心灵上的平静而已。先前有许多年,医生会建议做预防性乳房切除术,即便没有任何手术可以完全清除乳房组织,当时也没有确凿的数据证明这个方法拯救了一些人的性命。然而现在已经有证据证明,这种极端的做法的确降低了乳癌高危妇女的死亡率。以一项调查为例,在动了这手术的639人中,只有2人真的死于乳癌,而不是统计数字预期的20到40人。同样地,在40岁前(但在已经不再生育之后)切除卵巢,可降低卵巢癌和乳癌的几率。遗传分析赋予了妇女作决定的能力,而这真的可以造成生死差别。
但是,DNA分析使我们的未来也能有更多用比较温和的方法对抗乳癌的机会,如同密歇根的研究计划揭露的另一个故事。安妮的一位堂姐得知自己十之八九带有蹂躏其家族的BRCA1突变,由于她已经有多年没有照乳房X光片(讽刺的是,在高危家族里,这种由于恐惧而造成的疏忽并不罕见),于是她恐慌了。那天稍晚,韦伯安排她照乳房X光片,结果发现一个初期阶段的小肿瘤,它可以轻易切除,但是在例行检查中肯定会被忽略。自我检查和定期照乳房X光片无疑拯救了许多人的性命,但是就一些案例而言,推广这些检查的运动有可能造成出乎意料的后果,让这些人误以为自己很安全。对有遗传疾病风险的人进行筛检,让我们得以发现谁是那些在做影像检查时特别严格的人。风险愈高,就需要愈仔细的监测。长此以往,我们才可望在愈来愈少的海水中,找到愈来愈多的针。
南西·魏克斯勒来自亨廷顿氏症家族,安妮则来自乳癌家族,她们俩都属于可以利用新筛检法来了解其遗传命运的新一代。随着我们对比较常见的成人疾病,例如糖尿病和心脏病等的遗传原因更加了解后,生物科学的预测力量将会愈来愈强,让我们得知攸关我们每个人的遗传命运。
过去10年来,很少有疾病像阿尔茨海默氏症一样,让那么多人感到恐惧,每年因这种病而身心遭受折磨的人愈来愈多,现在有超过400万名美国人罹患这种病。患者的亲友首先会注意到他们犯了轻微的失忆,像是想不起来最近的事情或找不到正确的字眼,而他们可能会把这些情形归咎于年纪大了。接着患者的情绪可能有明显变化的迹象,但这在老年人身上也并不算不正常。但是随着病程发展,病人的症状会日益明显,让人不至于再弄错。丧失记忆的情形很快会变得异常严重,造成他们无法处理平时熟悉的工作,甚至简单的家事。语言愈来愈不流畅,经常说到一半就因为思绪中断而停下来。患者在知道自己的这些改变后可能会罹患忧郁症,使日益恶化的性格改变问题更加严重。重度阿尔茨海默氏症患者不知道自己是谁或身在何处,甚至认不出最亲近的人。随着记忆与性情不断恶化,他们所拥有的个人特质也逐渐被摧毁。
阿尔茨海默氏症一般在60岁左右开始发病,但有一种比较罕见,占所有病例5%左右的类型,会在40岁左右就发病。这种早发型阿尔茨海默氏症会让病患家属仿佛置身地狱,就跟亨廷顿氏症患者的亲友一样。早发型阿尔茨海默氏症患者在生命颠峰时期发病,然后逐渐遭到无情的摧毁。在数代当中有多位成员患病的家族,以前会被描述为遭到毁家灭门的“生物大屠杀”(biological Holocaust)。玛丽-克莱尔·金在她突破性的乳癌研究中,首度提出一个论点,认为任何疾病的早发形式可能比一般形式具有更清楚的遗传原因;早期大多数的阿尔茨海默氏症研究就是根据这个论点,把重心放在了早发型阿尔茨海默氏症上。到了1995年,研究人员已经发现三个相关的基因,它们都跟淀粉样蛋白(amyloid protein)沉积的处理过程有关,早在1906年,阿尔茨海默医生(Alois Alzheimer)首度描述这种疾病时,就已经提到病患脑部有淀粉样蛋白堆积的情形。因此早发型阿尔茨海默氏症显然是遗传的,但是其他比较常见的阿尔茨海默氏症类型呢?
杜克大学(Duke University)的罗塞斯(Allen Roses)没有跟着大伙走,反而选择研究常见得多的晚发型阿尔茨海默氏症。晚发型仅偶尔出现在家族中,例如,在1984年宣布患病的前总统里根,他在两年后失去了死于同一型阿尔茨海默氏症的哥哥尼尔(Neil),他们的母亲也是死于这一型。
罗塞斯是神经学家,对假肥大型肌营养不良症等肌肉疾病学有专长,他在1984年开始研究晚发型阿尔茨海默氏症。1990年,他宣称第19号染色体上的一个基因显然与这种病有关,但这个说法遭到质疑。最令罗塞斯高兴的事,莫过于证明别人都错了。两年后,他真的找出了这个重要的基因。最后发现它编码载脂蛋白E (apolipoprotein E, APOE),这种蛋白质与胆固醇的处理过程有关。这个基因有三种形式(即有三种等位基因),APOEε2、APOEε3和APOEε4,但是后来证明具有决定性角色的是APOEε4:只要有一份这个变异,一个人罹患阿尔茨海默氏症的几率就会增加四倍。若是有两份,则患病几率会比没有APOEε4等位基因的人高10倍。罗塞斯发现有两个APOEε4的人当中,55%会在80岁前发病。这种相互关系是否可以作为进行遗传检测的基础?可能不行。尽管与这个疾病有关,APOEε4等位基因仍很常见,要作为检测阿尔茨海默氏症的指标,它还不够准确。虽然有APOEε4等位基因的人罹患阿尔茨海默氏症的可能性较高,但还是有很多有两个APOEε4等位基因的人没有发病。但是运用APOEε4筛检,再配合临床评估,的确提高了诊断阿尔茨海默氏症的准确度。或许等我们了解了它们之间的因果关系后,就可以改良遗传分析技术。最近在实验鼠身上引发阿尔茨海默氏症的研究显示,APOE与造成阿尔茨海默氏症患者神经细胞死亡的蛋白质的代谢作用有关。
治疗方面呢?我们在治疗大多数的遗传疾病上,进展仍跟亨廷顿氏症一样令人沮丧。我们已经有足够的知识可以进行诊断,或许也能避免它们发生,但还没有找到治疗方法。幸好遗传学知识已让我们找到一些疾病的疗法,只可惜这些疗法中极少像苯丙酮酸尿症的治疗法一样简单有效——苯丙酮酸尿症患者可以通过限制一些饮食项目,恢复正常的生活。
遗传疾病经常造成特定组织的细胞逐一死亡,如假肥大型肌营养不良症的肌肉细胞、亨廷顿氏症和阿尔茨海默氏症的神经细胞。对于这种在不知不觉中恶化的疾病,目前仍没有权宜之法。虽然干细胞疗法目前仍在早期发展阶段,但我相信最终我们利用干细胞来治疗这类疾病的机会是很高的。人体内大多数的细胞只能自我复制,例如肝细胞只能制造肝细胞,但是干细胞可以制造多种类型的特化细胞。在最简单的例子中,一个刚受精的卵子(潜力最大的干细胞)最终可以制造出所有216种已知的人类细胞。因此干细胞最容易从胚胎中取得;它们也存在于成人体内,但成人的干细胞不像胚胎的干细胞能分化成任一类型的细胞。我们才刚开始了解如何诱导干细胞制造特定类型的细胞,希望未来我们可以用健康的新细胞替换亨廷顿氏症和阿尔茨海默氏症患者失去的脑细胞。但是我要提醒,我们仍须经过漫长的努力,才能彻底了解使一个细胞朝特定方向发展的分子启动机制。我们大约还要10年左右时间来解决这个发育生物学上的问题,才有办法妥善研究干细胞的治疗价值。如果这类研究因宗教考虑而受阻,这对科学及所有可能受益于干细胞疗法的人来说,都是一个悲剧。民意调查一贯显示,大多数美国人支持使用胚胎的干细胞来进行研究,然而政治人物却持续迎合少数大声反对的宗教团体,结果在美国形成立法加以限制,阻碍了这类具有极大潜在价值的技术发展。
就目前言,在遗传疾病的治疗上,我们还没有办法以干细胞疗法来更替大量的细胞,但是或许可以补充一种失去的蛋白质。发生率为四万分之一的高雪氏症(Gaucher disease)是一种罕见的疾病,起因于葡萄糖脑苷脂酶(glucocerebrosidase)的基因发生突变。葡萄糖脑苷脂酶协助分解一种特殊的脂肪分子,若是没有它们,这种脂肪分子会在人体细胞里堆积,造成伤害。高雪氏病有可能造成严重的伤害,其症状包括骨头疼痛与贫血。早在1974年,医界就开始尝试直接补充病人丧失的这种酶。结果证明这种疗法很有潜力,但它的准备工作却是一场恶梦:用来补充的酶必须从人类的胎盘中取得,而从两万个胎盘中取得的量,只够一个病人一年所需。在20世纪90年代初,研究人员有了重大突破,合成出这种酶的改良形式,让最需要它们的细胞更有效地吸收。1994年,基酶(Genzyme)生技公司开始运用DNA重组法制造这种改良型的酶。高雪氏症的治疗法不是解决造成这种病的遗传根源,而是反制这种突变所造成的效果,把缺陷基因无法提供的重要蛋白质提供给病人。
通过这种生化途径来矫正基因异常情况的做法,显然可行又有效。但是即使重组法的效率惊人,这种治疗法仍很昂贵,每年要17.5万美元,而且必须持续补充蛋白质,对病人来说也是一种负担。在这种情况下、遗传学家自然会希望能找到一种实际可行的方法来除去病根,而不是仅反制突变所造成的作用而已。遗传疾病的理想疗法是改变遗传,亦即矫正造成问题的基因。这种基因疗法可以让病患终生受益,一旦矫正好基因,就可以持续一生。至少在原则上有两种方法:体细胞基因疗法(somatic gene therapy),亦即改变病人体细胞的基因;生殖细胞疗法(germ-line therapy),亦即改变病患精子或卵子内的基因,防止有害的突变传给下一代。
以这类方法来解决遗传缺陷所造成的祸害,显然是很合理的做法,但是专业人士或一般民众对基因疗法的构想,却不是很能接受。其实这种反应也不足为奇:一个连对谷物进行基因改造都极其谨慎的文化,想当然会反对基因克隆人类(你也可以称之为基因改造人类[GM human]),无论其潜在好处有多大。这样的文化会对生殖细胞疗法更加反弹也是可以预期的,因为操控DNA有破坏基因的危险。在体细胞基因疗法中,损坏造成的效应可能有限;但在生殖细胞疗法中,这类损坏有可能意外造成残障。即便是支持这些疗法的人,包括我在内,也会在确保这种技术绝不会造成意外伤害后,才会建议实行。不过,许多科学家坚信我们永远不该尝试种系基因疗法。无论是基于道德观或对未知事物毫无缘由的恐惧,我认为这类论点终究无法令人信服。基本上,生殖细胞疗法只是把无意间出错的事物矫正回来。但是目前这在学术上仍有争议的原因是,生殖细胞疗法仍然远远超出我们现有的技术能力。在能掌握它之前,我们应该把重心放在发展体细胞基因疗法上,使其成为一个强大的工具。
第一个显然获得成功的基因疗法,是1990年美国国家卫生研究院的安德森(French Anderson)、布莱斯(Michael Blaese)和卡尔弗(Ken Culver)进行的。他们选择治疗的目标是一种非常罕见的疾病,称为腺苷脱氨酶(adenosine deaminase, ADA)缺乏症,病患的免疫系统因为缺乏一种酶而丧失功能,造成跟“泡泡男孩”维特一样无法抵抗疾病。他们的实验对象是两个小女孩,4岁的阿善蒂·狄西瓦(Ashanti DeSilva)以及9岁的辛迪·柯特薛尔(Cindy Cutshall)。
新基因要怎么注入病患体内?在当时,反转录病毒(retrovirus)似乎是合理的选择。一般来说,病毒是有效的基因载体,它们靠把自己的DNA注入其他的细胞来生存。反转录病毒是一群特殊的病毒,拥有的遗传物质是RNA,不是DNA。大多数的病毒在感染一个细胞后开始繁殖,然后杀死宿主细胞,“子代”的病毒逸出后再去感染其他的细胞。但是反转录病毒一般对宿主细胞比较仁慈和温和,新的病毒复本会自然离开,并不会摧毁宿主细胞。这并不是说反转录病毒对宿主生物体的伤害较小;有时恰恰相反,艾滋病毒HIV就是一个实例(HIV或许是最广为人知的反转录病毒)。但这的确代表病毒基因(以及这个病毒可能加载的额外基因)会永久成为未被摧毁的细胞基因组的一部分。遗传工程已能制造出足够安全的反转录病毒,供基因治疗使用;在除去病毒中所有将被入侵的宿主细胞基因组所不需要的基因后,反转录病毒就成为了理想的基因载体。
但是我们仍旧有个问题必须解决,亦即如何锁定那些受突变影响的细胞,也就是需要换装“替代基因”的细胞。如今这仍是基因疗法所面临的最艰巨挑战。如何把好基因植入肌肉细胞,以便治疗假肥大型肌营养不良症?如何把好基因植入肺细胞和脑细胞,以便治疗纤维囊泡症和亨廷顿氏症?因此,在首度进行基因疗法的实验时,以鲜为人知的腺苷脱氨酶缺乏症为目标,是很合理的选择。因为要治疗这个病症所需锁定的目标细胞,是在血液中循环的免疫系统细胞,很容易取得。安德森的研究小组从这两个女孩的血液中取得大量的免疫细胞,在培养皿中培养,再让它们接触携带了正常基因的反转录病毒。等这些细胞原有的DNA和携带替代基因的病毒基因组结合后,再把这些细胞重新注入病人的血液中。
1990年9月,阿善蒂率先接受了这个疗法;辛迪则是在4个月后。她们每几个月就注射一次经过基因改良的免疫细胞。同时,她们仍继续接受与基因无关的酶替代疗法,跟高雪氏症患者的疗法相同,但是剂量较少。这是美国国家卫生研究院的人类基因疗法小组委员会要求的预防措施,他们认为在没有任何安全保障的情况下让这两个女孩接受新疗法太过危险,这论点很合理。虽然这个实验是在不完全控制的情况下进行的,但似乎仍获得成效:两个女孩的免疫系统都有改善,而克服轻微感染的能力也变得较好。我个人可以作证,辛迪在1992年和家人造访冷泉港实验室时,看起来就像个非常健康的11岁女孩。然而在10余年后,这个实验仍无法获得完全确定的结果。现在阿善蒂的免疫系统功能正朝正常的水平前进,但她的T细胞(胸腺淋巴细胞)大约只有1/4是来自基因疗法。在辛迪的血液中,来自基因疗法的T细胞所占的比例更小,但现在她的免疫系统运作得很好。不过我们还是很难说这两个女孩的进展有多少是基因疗法的成果,又有多少是持续接受酶疗法的成果。这样的结果太过模糊,所以不能毫无保留地算是基因疗法的成功。
阿善蒂和辛迪的实验并不是美国国家卫生研究院第一次插手基因疗法领域的案例。事实上,该院的人类基因疗法小组委员会正是为了回应第一个基因疗法实验,而在1980年成立的。那次实验不仅失败,还引起争议,以至于美国政府几乎要扼杀掉这个才刚刚起步的领域。
根据各方说法,这个事件的主角是克莱恩(Martin Cline),一个聪明且雄心壮志的临床医生,向来致力于减轻病人的痛苦。他对β-地中海型贫血症特别感兴趣,也就是摩戴尔(Bernadette Modell)在伦敦的塞浦路斯族群中筛检出的血红素疾病。在动物实验成功后,克莱恩向他任职的加州大学洛杉矶分校审查委员会,申请使用非重组DNA (nonrecombinant DNA)进行基因疗法的人体实验。当他的申请仍在审查过程中时,过度热衷的克莱恩却已经安排对两名非美国境内的妇女(分别在以色列和意大利)进行实验,但他使用的是重组基因,当时国家卫生研究院的规定仍禁止使用重组基因。在回到洛杉矶后,克莱恩发现他的申请被驳回;审查委员会裁定必须要有更多动物实验的数据,才能批准人体实验。克莱恩几乎违反了所有的规定:他不仅在没有获得授权下就治疗人类,还使用了明确被禁止的方法。克莱恩也为此承受了苦果,他失去联邦经费,并被迫辞去系主任的职位。基因疗法也失去了它的第一个医生。
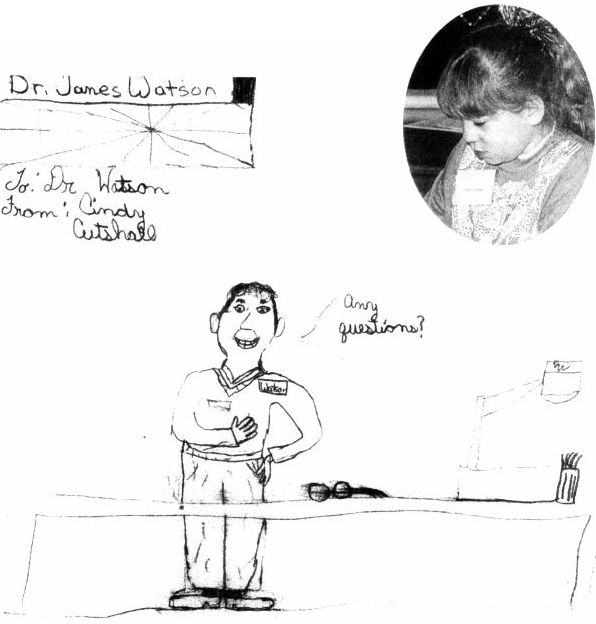
辛迪·柯特薛尔是最早参与基因疗法的病人。在到冷泉港实验室参观后,她给沃森寄了一张沃森演讲时的图画。
在克莱恩事件后,仍然有尝试基因疗法的科学家因违反规则而惹上麻烦。遗憾的是,一直要到一位尝试基因疗法的病人死亡,人们才深切地体会到,基因疗法这种涉及病毒、生长因子和病人的复杂治疗方法是有危险的。此外,由于基因疗法有许多未知因素,因此所有与人类有关的程序,绝对有必要接受严格的监督。亚利桑那州少年盖辛格(Jesse Gelsinger)之所以死亡,不仅是因为我们具备的知识尚不足以有充分把握来预测一个人对基因疗法的反应,也因为科学家走了不能原谅的快捷方式。
1999年,盖辛格听说宾州大学人类基因疗法研究所所长威尔逊(James Wilson)正在进行一个基因疗法实验。盖辛格罹患鸟氨酸氨甲酰转移酶(ornithine transcarbamylase, OTC)缺乏症,这种遗传疾病会破坏肝脏处理尿素的能力,而尿素是蛋白质代谢作用的自然产物。如果不接受治疗,这种病有可能致命。虽然它跟苯丙酮酸尿症一样,可以借由简单的药物治疗和适当的饮食来控制,但是OTC缺乏症的确会使病人特别容易罹患其他的疾病。18岁的盖辛格只是轻微病例,但是幼年一次险些因这病症而死亡的经验,让他壮着胆子自愿参与进临床实验,希望能为自己和其他的病患找到解药。宾州大学的基因疗法目标在于把腺病毒(adenovirus,引起普通型感冒的病毒之一)当做已矫正基因的载体,但是在把携带正常OTC基因的病毒注入盖辛格的肝脏数小时后,他开始发烧,接着发生严重的感染,伴随有血栓和肝脏出血。在注射三天后,盖辛格不幸死亡。
这名少年的病逝不仅对他的家人是一大打击,也令研究圈震惊。详尽的调查发现,这当中有严重的程序缺失,最显著的错误是:虽然先前在相同的研究中已有两名病患出现肝中毒的症状,但这些病例却没有通报给任何管制当局,也没有告知自愿参与研究的患者。如果盖辛格家知道这点,他或许不会这么急着自告奋勇,说不定他至今仍活着。这场悲剧对基因疗法的进展是一大打击。有一阵子,美国食品及药物管理局要求全美国的大学及其他数个研究计划终止这类实验。参议院惟一的医生,来自田纳西州的佛瑞斯特(Bill Frist)也深入调查了人体实验的通报程序;克林顿总统则要求改进“告知同意”的标准,认为实验对象有权获知所有的潜在风险。如果盖辛格的事件带来任何正面效果,那就是联邦政府对人体实验的监督愈来愈严格。
就在基因疗法界仍因盖辛格死亡事件的冲击而震惊不已时,法国传来令人振奋的成功消息。法方锁定的目标是严重综合型免疫缺乏症(SCID),亦即造成维特终生以“泡泡”为家的疾病。虽然骨髓移植可以达到治疗效果,但成功率只有40%左右,即使移植成功,也经常有可怕的并发症,如同维特不幸的案例。2000年,费希尔(Alain Fischer)在巴黎内克医院(Necker Hospital)领导的医疗小组,对两名婴儿进行了基因疗法,他们跟维特一样,也是一出生就住在无菌的隔离空间里。这个小组采取跟治疗腺苷脱氨酶缺乏症相同的方法,以反转录病毒为载体,把需要的基因送入取自婴儿的细胞,再把这些细胞重新注入婴儿体内。但法国小组作出了一个相当创新的改变,他们是从婴儿的骨髓中取得的待改造细胞。这个方法运用的是骨髓的免疫干细胞,而不是一般在血液中找到的普通T细胞,如果成功的话,这个方法可以成为自发永续的基因疗法。干细胞在复制时,不仅本身的数目会增加,由它们自然分化形成的特化体细胞(specialized somatic cell)也会增多。因此,由经过改造的干细胞所产生的T细胞,也会携带相同的新插入基因,患者就不必持续注射改造过的细胞。
事情也如预期发展:10个月后,两名病童身上都能找到含有一份正常基因的T细胞,而他们的免疫系统功能也跟正常儿童一样。费希尔的疗法后来也被用于其他的SCID病童。在经过漫长而且一波三折的开端后,基因疗法终于获得明确的成功。但是这场香槟庆祝会的气氛没有持续多久。2002年10月,医生发现最早接受这种治疗的两个病童中,有一个患了白血病,这种骨髓方面的癌症会使特定类型的细胞生产过剩。尽管目前为止无法确认这是否是基因疗法所造成的,但是间接证据仍然很有说服力。基因疗法似乎治好了这名婴儿的SCID,却引起白血病的副作用。
副作用向来是医学难以摆脱的问题。药物可能不只对原先锁定的目标造成影响,而手术则可能会引起并发症。虽然在许多方面,基因疗法都与传统医学不同,但现在我们已经知道它仍摆脱不了发生意外结果的定律。费希尔的SCID疗法可能在修正原有基因的过程中,无意间产生了新的问题。毕竟,任何把病毒DNA插入病人细胞DNA的疗法,原本就潜藏着风险,因为这个外来DNA有可能偶然地阻断某个重要基因的功能。由于发生这种情况的细胞通常都会死亡,因此这样的意外一般不会造成影响。但是这个遭阻断的基因在丧失功能后,其后果有可能不是导致细胞死亡,而是使细胞毫无节制地增殖;这也就是说,病毒基因插入法有可能引起癌症。这似乎就是那名SCID宝宝发生的情况。
看来基因疗法似乎还要很长的时间,才能创造基因革命开始时预见的奇迹。盖辛格死亡事件是一次严重的失败,但是SCID疗法的白血病副作用造成的伤害甚至更大。在盖辛格的个案中,主因在于不可原谅的管理疏失,这个问题通过更严格的规定应该已经解决了。但是副作用的问题至今还没有可行的解决方法。在这个例子中,或许我们只能无奈地自我安慰:至少基因疗法治好了SCID,起码SCID是比这疗法引起的白血症还严重的病。幸好那名罹患白血病的男婴对化学疗法的反应显然良好。然而,盖辛格和白血病的事件具体呈现出了许多困难的问题,如果体细胞基因疗法要成为医学主流,就必须先解决这些问题。而我也不会天真地以为,未来的实验就不会遇到更多的困难。或许还要一段时间,我们才能确切地宣称已经解决掉所有想像得到的危险。尽管如此,我仍旧认为,这个技术破解遗传疾病宿命的潜力实在很大,医学界绝不能放弃它。

目前还是好消息:费希尔(左)和他的合作伙伴卡瓦沙那-卡佛(Marina Cavazzana-Calvo)在2000年4月宣布他们突破性的基因疗法获得了成功。
你的DNA可以告诉别人很多有关你的事。如同先前所述,如果你的家族有亨廷顿氏症的病史,你的DNA可以预示你的未来;不久后,DNA或许也可以指出你罹患心脏病等某种常见致命疾病的相对风险,视你是否拥有某个(或数种)特殊的变异基因而定。你拥有哪一型的APOE基因,已经可以作为阿尔茨海默氏症的指标。但是我们该不该担心,这个极度隐私的信息有一天可能会成为对你不利的工具?对许多美国人来说,他们最担心的无疑是有一天遗传信息会让他们无法参加健康保险。
2000年,《美国人类遗传学期刊》(American Journal of Human Genetics)发表了一项调查结果,这项调查询问健康保险业者,如果可以获得遗传信息的话,他们是否会根据遗传信息来调整保费的费率?他们会不会打算要求健康状况良好,但因基因突变而容易患病的客户付更高的费用?大约有2/3的受访者坦承会,而剩下的1/3十之八九在说谎。保险公司不是慈善机构,而是生意人,必须让股东满意。我们没有理由假定,他们在可以自行其是的情况下会改变向来的做法,亦即把有风险的客户保费提到最高,而尽量避免替最可能申请理赔的客户承保。这份报告也引述了一个案例:有一家保险公司怀疑一位客户患有遗传疾病而提高其保费费率,只因为这人曾经申请做亨廷顿氏症的诊断检测。
随着我们对自身的分子构造日益了解,在这场基因赌博中不幸抽到下下签的人是否势必得付出某种代价?我们凭什么认为这类歧视待遇仅限于保险业者?我的DNA可能显示我有罹患心脏病、中风、酗酒或忧郁症的风险,这类信息会不会让雇主在雇用我以前三思?
从这类问题看来,在赫胥黎想像的25世纪来临前,他笔下的《美丽新世界》(Brave New World)就已然堂堂登场了。DNA已经深入21世纪的生活,就像一个无法再关回瓶中的精灵。至于我们要如何运用它,决定权则在于我们这个民主社会。不幸的是,在民主社会中,立法的速度似乎总是跟不上实际的需求:一般而言,通常要有数起车祸发生后,某个危险的十字路口才会装上红绿灯。可能要到有一些极端不公平的可怕事件发生,有人因自己的基因组而成为受害者之后,才会有推动适当法律通过的动力。这应该是什么样的法律昵?“基因隐私”应该是基本准则,但不一定是最终目标。基因隐私应该与社会的其他优先事项达成平衡,尤其是在对抗疾病上。在这方面要有所进展,医学研究人员能否尽量取用大众的基因资料显得日益重要。立法不应阻挡我们充分开发DNA的诸多潜力,包括减轻人类痛苦、告诉人们有关其本身及祖先之事,或协助找出罪犯等等,而且它至少要能确保,所有人民不会因其基因而被剥夺民权或人权。
同时,或许我们可以欣慰地得知,尽管保险业已经有大量可以自由使用的基因资料,而且无论他们给民意调查的答案是什么,整体来看,保险公司至今似乎还不会冲动地把遗传基因列为保险费率的计算因子。我遗传到的皮肤极白,现在也已证明这样的肤色容易得癌症,但我上次检视保单时,并没发现我的保费因为这个因素而增加。同样地,这当中的原因并不在于心怀慈善,而在于商业考虑。保险业者一向利用精算表来设定保险费率,这种精算表则是根据我们的生活方式来估算整体的健康状况与寿命。我想就算基因数据到处都可取得,保险业者还是会发现生活方式的因素,例如一个人抽不抽烟,是在煤矿还是花店工作等等,更能预测一个人的健康风险,远胜过绝大多数由遗传变异所决定的细微个人差异。那些从DNA得知自己未来的健康必然会逐渐衰退的人,无疑需要法律的特别保护。但是容易患心脏病和癌症的体质势必会很普遍也很复杂,因此要以它们作为缩减成本的基础并不切实际。所谓保险就是以大多数没有病痛、不必申请理赔的人所付的保费,来救助少数不幸者,这样的基本前提不太可能因为获得了更多遗传信息就废除掉。
尽管个人的权利不会因为DNA所揭露的信息而受到侵害,但在心灵上可能无法这么容易恢复平静;如同南西·魏克斯勒深切体会到的,基因认知可能会揭露可怕的未来。我同意她的看法:知道一些我们无力补救或改善的事情,一点意义也没有。阿尔茨海默氏症是我这个年纪的人最担忧的事情之一,但是在缺乏经过验证的疗法下,我一点也不想检验我是否有APOEε4等位基因。温特确定拥有一份APOEε4,我们之所以知道,是因为他坚持公开宣布赛雷拉基因公司所定序的基因组是他的。尽管这个等位基因在胆固醇处理过程中具有一定的作用,但它不仅会使人患阿尔茨海默氏症的几率提高,连患心脏疾病的几率也会提高。(在任何方面,APOEε4都不是一项有利资产。)在得知这个基因事实后,温特明智地尽量釆取预防措施:他服用一种叫做他汀(statin)的药物,这种药可以降低胆固醇含量,而且或许可以阻碍或预防阿尔茨海默氏症的发生。即使我不知道自己的APOE等位基因情况,但我也在服用他汀,我的想法是反正采取一些预防措施也没什么不好。如果他汀真的如药厂所宣称的那么有效,我们就可以多看几年温特引起的争议(希望也会有沃森引起的争议)。
只要我们一直停留在目前的“中间阶段”,亦即大致上只有诊断能力而没有治疗能力,那么基因知识仍会显得可怕。但这并不是我们第一次遇到这种医学困境。在20世纪初也有类似的情况,当时被诊断出糖尿病的婴儿等于被宣判死刑。如今随着胰岛素疗法的问世,这类儿童可以好好地长大成人。我们作研究的目的就在于希望不久的将来,对于亨廷顿氏症这类疾病的诊断能有相同的结局:从宣判死刑变成开立疗方。
今日我们在处理遗传疾病时,能做的已经比20年前多得多;例如唐氏症和纤维嚢泡症患者的预期寿命不断增长,就是医学进展的证明。但是目前我们最强大的武器仍是早期的诊断。至于要不要接受筛检,最好是留给个人或父母来抉择,因为他们必须直接承受这些基因信息所带来的负担。就产前诊断而言,则应由孕妇来作决定。这并不是说他人不用参与,而是说最终的选择权应该归于孕妇,不仅因为她是胎儿的母亲,也因为无论我们喜不喜欢,现今的世界仍是由妇女来承担日常照顾儿童的责任。然而,无论作决定的各种考虑为何,对我来说有一件事是很明确的:长久以来,遗传疾病已经给无数家庭带来无法想像的悲惨后果,凯萝·卡利饱受亨廷顿氏症折磨的家庭就是一例。筛检可以借由预防遗传疾病的发生来减少这类不幸。在发展出筛检方法后,不把它们告知那些可能想采用的人是不合乎良知的,而不让所有人都能采用它们,更是令人无法原谅的做法。
这部迪斯尼影片的结局是,最后泡泡男孩竟然很健康。
唐恩最初基于这些特征而称这种病为“蒙古症”(mongolism),他1866年发表的论文即名为《白痴的种族分类观察结果:(Observations of an Erthnic Classification of idiots)。由于赞同当时种族主义者的进化观点,他认为唐氏症是从高贵的高加索人神(白种人)往“劣等的”蒙古人种(Mongoloid)退化的情形。不过他也在结论中提到,他所谓的“退化”(retrogression)使那些坚信白种人与非白种人不是同一物种的论点,愈发站不住脚。
在英国,被诊断出有唐氏症的胎儿中,有92%会遭到堕胎。一般而言,只有愿意考虑堕胎的人才会接受产前筛检(如果无论筛检结果为何,母亲都打算生下胎儿的话,就没有理由冒险做可能伤害胎儿的检测),因此这个数字注定会偏高。
这个年轻人的父亲有1/2的几率会从祖父遗传到突变,如果真的遗传到,他把突变传给儿子的几率也是1/2。因此儿子得到突变的几率是这两个独立事件的几率乘积,亦即1/4。
70%的数字适用于北欧血统的人,纤维囊泡症在北欧血统中最常见。然而在非洲裔和德系犹太裔的美国人当中,△F508只占纤维囊泡症突变的35%左右。这类血统上的差异使筛检计划的设计变得很复杂。